Plaquetas, cuagulacion y fibrinolisis por Juan F. Martinez 84445
-Activacion Plaquetaria: Esta primera imagen nos muestra como se lleva a cabo activacion plaquetaria: -Primero tiene que haber una lesion, donde el factor vW procede a adherirse al colageno expuesto en la lesion endotelial. - segundo la adhesion: las plaquetas se adhieren al factor vW via el receptor GpIb en el lugar de la lesion unicamente. Las plaquetas comienzan a liberar ADP y Calcio necesario para la cascada de cuagulacion. El ADP ayuda a que las plaquetas se adhieran al endotelio. -tercero la activacion: el ADP enlazado al receptor induce la expresion de GpIIb/IIIa en la superficie de las plaquetas. -cuarto: agregacion: el fibrinogeno se adhiere al receptor GpIIb/ IIIa y enlazan plaquetas.
-Coagulacion: En cuanto a la cuagulacion sabemos que esta tiene dos vias la via intrinseca y la via extrinseca. La via extrinseca consiste de los factores VII que es activado por el factor tisular para formar VIIa que a su vez escinde al factor X en Xa, que a su vez va a escindir el factor II (protrombina) en IIa (trombina) que a su vez escinde al fibrinogeno para formar los monomeros de fibrina que forman la red de fibrina que actua para estabilizar el tapon de plaquetas. Por otra parte la via intrinseca consta del factor XII que es activado por colageno, membrana basal o plaquetas activadas y escindido a XIIa que a su vez escinde al factor IX en IXa que a su vez es escindido por el factor VIIIa y de esta forma el factor IXa escinde al factor X y se conectan ambas vias para obtener el mismo resultado la red de fibrina y lo que es la cuagulacion.
-Fibrinolisis y sistema fibrinolitico: La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. Este diagrama nos resume y nos dice que esta se puede llevar a cabo por diversas vias. La activación de plasmina a partir de plasminógeno puede ocurrir a través de las vías, extrínseca, intrínseca, exogena y endogena: En la vía extrínseca se produce una segregación de diversas sustancias que posibilitan la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA. En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno que activa la plasmina y degradan los productos de la fibrina. No esta demás mencionar que la antitrombina inhibe las formas activadas de los factores II, VII, IX, X, XI y XII.
El proceso de agregación plaquetaría se desarrolla en dos secuencias: En primer lugar, mediante la adhesión de las plaquetas a la superficie vascular lesionada (subendotelio) y posteriormente, mediante la activación plaquetaría, cuyo resultado final consiste en la formación de un entramado de plaquetas y cadenas de fibrinogeno que forma el trombo.
La activación plaquetaria comprende: -la formación y liberación de sustancias vasoactivas o agonistas (tromboxano A2, adenosina trifosfato, trombina,epinefrina, serotonina y colageno) que favorecen al proceso de agragacion induciendo a su vez la activacion de otras plaquetas mediante una reaccion de cascada. - la activacion de receptores de proteinas en la membrana plaquetaria, entre los que se encuentra el responsable de la fijación de las plaquetas a la zona lesionada (factor de von willerbrand) y el mas importante, el receptor glucoproteinas IIb\IIIa que reconoce y fija las cadenas de fibrinogeno, formando la trama final del tapon hemostatico.
-Factor de la coagulación I, Se convierte en fibrina por acción de la trombina. La fibrina constituye la red que forma el coágulo.en el plasma xiste una cantidad de 250-400mg\dl. -Factor de coagulación II tambien conocido como prototrombina. Se convierte en trombina por la acción del factor Xa. La trombina cataliza la formación de fibrina a partir de fibrinógeno.en el plasma existe en una cantidad de 10-14mg\dl. -factor III o factor tisular de trmboplastina. Se libera con el daño celular; participa junto con el factor VIIa en la activación del factor X por la vía extrínseca. -factor IV su nombre es ion calcio, Media la unión de los factores IX, X, VII y II a fosfolípidos de membrana. Su nivel en el plasma es de 4-5mg\dl.
El trombo es un coágulo sanguíneo que se forma en un vaso y permanece allí. La embolia es un coágulo que se desplaza desde el sitio donde se formó a otro lugar en el cuerpo. El trombo o embolia puede producirse en un vaso sanguíneo y obstruir el flujo sanguíneo en ese lugar, impidiendo el suministro de oxígeno y flujo sanguíneo a los tejidos circundantes. Esto puede ocasionar un daño, destrucción (infarto) e incluso la muerte o necrosis de los tejidos que se encuentran en esa área.
Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida. Un coágulo sanguíneo que se forma dentro de una de las venas o las arterias se denomina trombo y también se puede formar en el corazón. Un trombo que se desprende y viaja desde un lugar en el cuerpo a otro se llama émbolo. Un trombo o émbolo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo. Una obstrucción en una arteria puede impedir que el oxígeno llegue a los tejidos en esa área, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede provocar daños en los tejidos o la muerte. Una obstrucción en una vena generalmente provocará acumulación de líquido e hinchazón. La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno. La fibrinolisis es un encadenamiento (proceso) enzimático compuesto por una serie de activadores e inhibidores los cuales regulan la conversión del proenzima circulante, plasminógeno, en el enzima activo plasmina. La producción de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina y esto es muy importante para el mantenimiento de la permeabilidad vascular . los componentes de la fibrinolisis son: plasminogeno, plasmita, activador tisular del plasminogeno, estreptoquinasa, uroquinasa, factor XII, precalicreina y quiminogeno de alto PM, PAI, glicoproteina rica en histidina, antiplasminas y a 2-antiplasmina.
Ana M. Igartua 86978 Activacion plaquetaria La sangre circula por el interior de los vasos, los glóbulos rojos (la más abundante masa celular sanguínea) lo hace por el centro del vaso, mientras que las plaquetas circulan más próximas a la superficie endotelial para vigilar la integridad del endotelio. El endotelio normal proporciona una superficie no adhesiva para las plaquetas. Cuando se rompe el endotelio, el colágeno expuesto favorece la adhesión de las plaquetas por medio de la interacción con una cantidad de receptores, como vemos en el video, pero es esencial para concretar la adhesión el Factor de Von Willebrand. A partir de esto y con el aumento del Ca++ intracelular se produce la activación de las plaquetas, lo que genera una serie de eventos que favorece la agregación plaquetaria, como son: -Cambio de forma y de ser una esfera regular pasa a tener prolongaciones. -Liberación del contenido de los gránulos (degranulación): ADP que torna a las plaquetas pegajosas,TX A2 derivado del Ácido Araquidónico (vasoconstrictor y agreganteplaquetario) actor II (trombina). Adhesión, Activación y Agregación son las fases que participan en la formación del tapón plaquetario . Proceso de coagulacion Los factores de coagulación son proteínas de la sangre que controlan el sangrado. Cuando un vaso sanguíneo se lesiona, sus paredes se contraen para limitar el flujo de sangre al área dañada. Entonces, pequeñas células llamadas plaquetas se adhieren al sitio de la lesión y se distribuyen a lo largo de la superficie del vaso sanguíneo. Al mismo tiempo, pequeños sacos al interior de las plaquetas liberan señales químicas para atraer a otras células al área y hacer que se aglutinen a fin de formar lo que se conoce como tapón plaquetario. En la superficie de estas plaquetas activadas muchos factores de coagulación diferentes trabajan juntos en una serie de reacciones químicas complejas (conocidas como cascada de la coagulación) para formar un coágulo de fibrina. El coágulo funciona como una red para detener el sangrado. Los factores de la coagulación circulan en la sangre sin estar activados. Cuando un vaso sanguíneo sufre una lesión se inicia la cascada de la coagulación y cada factor de la coagulación se activa en un orden específico para dar lugar a la formación del coágulo sanguíneo. Los factores de la coagulación se identifican con números romanos (e. g. factor I o FI). Fibrinolisis La fibrinólisis primaria se refiere a la descomposición normal de los coágulos, mientras que la fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa. Consecuencias Cuando se produce demasiado rápidamente, puede provocar hemorragias espectaculares (fibrinólisis hemorrágica) intra o postoperatorias (particularmente en Cirugía Torácica), o después de un parto, de un aborto o de un choque traumático; o bien puede provocar hemorragias menos importantes (hematuria) en el curso de ciertos cánceres (próstata, páncreas, estómago), de las cirrosis y de las leucosis. Obedece a la liberación de activadores de la profibrinolisina, o bien a la producción de un fermento proteolítico por los tejidos enfermos.
El proceso de formación del trombo es la consecuencia patológica de la activación del mecanismo hemostático, el cual depende de un sistema enzimático complejo regulado por la acción de diversos factores activadores e inhibidores. En su desarrollo están implicados los elementos circulantes, las estructuras de la pared vascular y las proteínas plasmáticas. El proceso de agregación plaquetaria se desarrolla en dos secuencias: en primer lugar, mediante la adhesión de las plaquetas a la superficie vascular lesionada (subendotelio); y, posteriormente, mediante la activación plaquetaria, cuyo resultado final consiste en la formación de un entramado de plaquetas y cadenas de fibrinógeno que forman el trombo. Por su parte, la activación de las plaquetas comprende: a) la formación y liberación de sustancias vasoactivas o agonistas plaquetarios (tromboxano A2, adenosina difosfato, trombina, epinefrina, serotonina, y colágeno) que favorecen el proceso de agregación induciendo a su vez la activación de otras plaquetas mediante una reacción en cascada b) la activación de receptores de proteínas en la membrana plaquetaria, entre los que se encuentra el responsable de la fijación de las plaquetas a la zona lesionada (factor de Von Willebrand) y el más importante, el receptor glucoproteína IIb/IIIa que reconoce y fija las cadenas de fibrinógeno, formando la trama final del tapón hemostático. Fisiológicamente, todo este proceso puede ser a su vez inhibido por otras sustancias, principalmente prostaciclina y óxido nítrico, que aumentan las concentraciones de AMP-cíclico en las plaquetas y en la pared vascular mediante una estimulación de la adenilciclasa (prostaciclina) o bien, aumentan las concentraciones de GMP-cíclico por estímulo de la guanilciclasa (óxido nítrico)
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden lavasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre. Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional. El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimáticopor el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse. Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas.
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc. En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares. A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos. Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas. Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada. Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola. Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
La cascada de coagulación se divide para su estudio, clásicamente en tres vias: La via intrínseca, la vía extrínseca y la vía común. Las vías intrínseca y extrínseca son las vías de iniciación de la cascada, mientras que la vía común es hacia donde confluyen las otras dos desembocando en la conversión de fibrinógeno en fibrina. Esta división es un tanto arbitraria y tiene más que ver con las deficiencias de las técnicas que en su momento se utilizaron para desentrañar los mecanismos implicados, que con lo que ocurre realmente en una lesión vascular; ya que en este último caso se establecen varias interrelacciones entre las vías de iniciación.
Cada reacción de estas vías da como resultado el ensamblado de un complejo compuesto por una enzima (factor de coagulación activado), un sustrato (proenzima de un factor de coagulación) y un cofactor que actúa posibilitando la reacción. Estos componentes se ensamblan en general sobre una superficie fosfolipídica y se mantienen unidos por medio de puentes formados por iones Ca2+. Por lo tanto la reacción en cascada tiende a producirse en un sitio donde este ensamblaje puede ocurrir; por ejemplo sobre la superficie de plaquetas activadas. Tanto la vía intrínseca como la vía extrínseca desembocan en la conversión del factor X en Xa (la letra "a" como subíndice "a" significa "activado") punto en el que se inicia la vía común.
Recibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso hemostático. El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular. En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación. En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como la celulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción. A esta vía es posible subdividirla en tres pasos: Recibió este nombre debido a que fue posible notar desde un primer momento que la iniciación de esta vía requería de factores ajenos a la sangre. Cuando la sangre entra en contacto con tejidos lesionados o se mezcla con extractos de tejidos, se genera muy rápidamente factor Xa. En este caso la activación de la proenzima X es mediada por un complejo formado por factor VII, Ca2+ y factor tisular unido a fosfolípidos provenientes de las membranas celulares rotas y de las plaquetas (antiguamente este complejo factor tisular-fosfolípidos era conocido como tromboplastina). El factor tisular es una lipoproteína sintetizada en el endotelio de los vasos sanguíneos de todos los tejidos, aunque es especialmente abundante en pulmón, cerebro y placenta. El factor tisular se encuentra normalmente "secuestrado" en el interior de las células endoteliales y es secretado en respuesta a una lesión, o bajo el efecto de algunas citoquinas tales como el Factor de Necrosis Tumoral (TNF), InterLeucina 1 (IL-1); o por endotoxinas bacterianas. La vía extrínseca es muy rápida, se cumple en apenas unos segundos y comprende dos pasos; mientras que la intrínseca insume varios minutos.
Coagulo y trombo Trombo: es una masa intravascular o intracardíaca formada por elementos de la sangre e insolubles en ella, se caracterizan por estar fijos a la pared del vaso por una zona donde son más rígidos y consistentes. Pueden ser arteriales o venosos.
Coagulos: intravasculares son postmorten son gelatinosos, con una porción declive rojo oscura porque los eritrocitos sedimentan por acción de la graverdad y un sobrenadante amarillo en grasa de pollo, no estan fijos a la pared del vaso. En cambio los trombos son mas consistentes, estan anclados en un punto a la pared del vaso o cardiaca, los arteriales consistentes de color blanco grisaceo y friables, formados por una red de plaquetas fibrina y leucocitos en degeneración (se generan en zonas donde hay turbulencia, como en la bifircación de los vasos) lo venosos son rojos y se desarrollan en zonas de estasis sanguineo por lo que retienen mayor número de hematíes, por eso son rojos
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón. La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca: • En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA. • En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y elinhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
El sistema fibrinolítico va a jugar además un papel importante en diversas situaciones en las que se produce proteolisis tisular, tales como inflamación, invasión tumoral o neovascularización (3, 4). La importancia fisiopatológica del sistema fibrinolítico deriva de que las alteraciones que condicionan un defecto de actividad fibrinolítica pueden predisponer a la trombosis, mientras que un exceso de activación favorecería la aparición de hemorragia. Además va a tener una gran importancia en el área terapéutica por la aplicación creciente de fármacos fibrinolíticos en el tratamiento de la trombosis, especialmente en el infarto agudo de miocardio (IAM). Principales componentes del sistema fibrinolítico: La fibrinolisis es un encadenamiento (proceso) enzimático compuesto por una serie de activadores e inhibidores los cuales regulan la conversión del proenzima circulante, plasminógeno, en el enzima activo plasmina. La producción de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina (5) y esto es muy importante para el mantenimiento de la permeabilidad vascular El plasminógeno o profibrinolisina es una glicoproteína sintetizada por el hígado, presente en el plasma sanguíneo y la mayor parte del fluido extracelular como el precursor inactivo de una enzima proteasa llamada plasmina. El plasminógeno es el componente central del sistema fibrinolítico del organismo, es decir, en la disolución de coágulos sanguíneos de craneados y otros organismos eucariotas. En humanos, la concentración de plasminógeno es entre 1.5 a 2 µmol/l.
Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro,1 derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos.
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre. Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo. La función plaquetaria consiste en el mantenimiento del corazón; esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio.
La Activación plaquetaria consiste cuando la superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP.
La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria(plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
El proceso de coagulación implica una serie de reacciones que intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares. A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (preacelerina factor V, protrombina Factor II, proconvertina factor VII, factor antihemofílico beta IX, factor Stuart X, tromboplastina plasmática XI y factor Hageman XII) son zimógenos sintetizados en el hígado.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada. Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
El fibrinógeno (factor I) es una glicoproteína compuesta por seis cadenas polipeptídicas: dos A-alfa, dos B-beta y dos gamma; unidas entre sí por puentes disulfuro.
Durante la fibrinolisis, después de que el coágulo se ha establecido, comienza la reparación de los tejidos afectados con el proceso de cicatrización. Para hacer posible esto el coágulo es colonizado por células que formarán nuevos tejidos y en el proceso va siendo degradado.
La degradación de la fibrina (fibrinólisis), componente mayoritaria del coágulo, es catalizada por la enzima plasmina, una serina proteasa que ataca las uniones peptídicas en la región triple hélice de los monómeros de fibrina. La plasmina se genera a partir del plasminógeno, un precursor inactivo; activándose tanto por la acción de factores intrínsecos (propios de la cascada de coagulación) como extrínsecos, el más importante de los cuales es producido por el endotelio vascular. Se le denomina "activador tisular del plasminógeno" (t-PA).
El gen de este factor ha sido clonado y actualmente se puede obtener la proteína producida por tecnología de ADN recombinante. Este factor suele utilizarse en clínica para favorecer la disolución de trombos.
El sistema de la coagulación mantiene la integridad vascular limitando la hemorragia y remodelando el vaso dañado, una vez que los mecanismos de reparación tisular han cesado. La hemostasia es el conjunto de mecanismos fisiológicos que se encargan de detener la hemorragia cuando se produce una injuria o daño vascular, es decir que existe un equilibrio en la necesidad fisiológica de mantener la sangre líquida permitiendo su circulación y cumplir sus funciones de transporte de oxígeno y elementos nutritivos a los tejidos y por otra parte la capacidad de detener el flujo cuando ocurre injuria vascular. ¿Quiénes conforman el sistema hemostático? 1- Vasos sanguíneos 2- Plaquetas 3- Mecanismo de la coagulación plasmática 4- Mecanismo fibrinolisis 5- Inhibidores o reguladores fisiológicos ¿Cuál es la secuencia cuando se rompe un vaso sanguíneo? 1. reacción o fase vascular (modificadores del tono vascular) 2. Reacción o fase plaquetaria (adhesión y agregación plaquetaria con la formación del trombo hemostático primario) 3. Activación de la coagulación sanguínea (trombo hemostático definitivo) 4. Activación de la fibrinolisis (digestión del trombo, recanalización del vaso)
EQUILIBRIO ENTRE LA COAGULACIÓN Y FIBRINÓLISIS La trombina tiene un efecto anticoagulante local, al conformar un complejo con la trombomodulina sobre la superficie endotelial, inhibe el coágulo sanguíneo por activación de la proteína C. La proteína C (proteína k dependiente), inicia la fibrinólisis por activación del plasminógeno y neutralizando un inhibidos activador del plasminógeno (PAI-1).
Las antitrombinas son inhibidores de la trombina y otras proteasas de la coagulación. La Antitrombina III es el más importante y posiblemente el causante del 50% de la fluidez natural de la sangre. La acción de la antitrombina III es catalizada por la heparina y por otras enzimas de la cascada de la coagulación como los factores Xa, XII, XI y X. Una deficiencia de antitrombina III se encuentra asociada a un aumento de trombosis.
La trombina tiene un efecto anticoagulante local, al conformar un complejo con la trombomodulina sobre la superficie endotelial, inhibe el coágulo sanguíneo por activación de la proteína C. La proteína K dependiente), inicial la fibrinólisis por activación del plasminógeno y neutralizando un inhibidor del plasminógeno (PAI-1)
FORMACIÓN DE FIBRINA Y FIBRINÓLISIS La introducción de la terapia a base de t-PA (tejido activador del plasminógeno) en la práctica clínica, ha puesto de presente la importancia de la fibrinólisis en el mantenimiento y función de la pared arterial. La formación de fibrina es regulada por un proceso enzimático de degradación que convierte la fibrina en fibrinógeno por acción de la plasmina. La plasmina está formada a partir de plasminógeno una globulina-b sintetizada por el hígado. La actividad de la plasmina está determinada por activadores o inhibidores del plasminógeno o inhibidores de la plasmina. La fibrinólisis es iniciada por el factor XIIA o uroquinasa activadora de plasminógeno (vía intríseca) o por un activador del plasminógeno (t-PA) vía intrínseca.
Los estrógenos disminuyen una glicoproteína rica en histidina y puede aumentar la fibrinólisis. Las progestinas están asociadas con una aumento en la actividad del plasminógeno.
Clínicamente como marcadores de trombosis se pueden utilizar el fibrinógeno plasmático, el tiempo de protrombina (indirectamente mide el factor VII) y el PAI-1. Recientemente niveles elevados de los fragmentos 1.2 (derivan de la conversión de protrombina o trombina). El PTT es de poco valor. El fibrinopéptido A y B así como varios productos de degradación del fibrinógeno, cambian en respuesta a la terapia hormonal y pueden medir indirectamente la aceleración de la coagulación. La dosificación de proteína C, S y posible a-antiplasmina, miden la acción de la Antitrombina III, en casos de deficiencia de esta proteínas puede ser útil para prevención de la trombosis, pero no informa sobre el riesgo de la inapropiada formación del trombo.
trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro,1 derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos. Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva. Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebro vascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores. Cualquier anormalidad o enfermedad de las plaquetas se denomina trombocitopatía,2 la cual puede consistir, ya sea en tener un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Se pueden producir desórdenes que reducen el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos. Sin embargo, otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragias. Las plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular; Estos dos factores de crecimiento han demostrado jugar un papel significativo en la regeneración y reparación del tejido conectivo; Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.
trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro,1 derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos. Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva. Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebro vascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores. Cualquier anormalidad o enfermedad de las plaquetas se denomina trombocitopatía,2 la cual puede consistir, ya sea en tener un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Se pueden producir desórdenes que reducen el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos. Sin embargo, otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragias. Las plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular; Estos dos factores de crecimiento han demostrado jugar un papel significativo en la regeneración y reparación del tejido conectivo; Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.
youel diaz 86760 Las plaquetas son producidas en el proceso de formación de las células sanguíneas llamado (trombopoyesis) en la médula ósea, por fragmentación en los bordes citoplasmáticos de los megacariocitos. El rango fisiológico de las plaquetas es de 150-400 x 109/litro. Alrededor de 1 x 1011 plaquetas de media son producidas cada día por un adulto sano. La expectativa de vida de las plaquetas circulantes es de 7 a 10 días. La producción de megacariocitos y plaquetas está regulada por la trombopoyetina, una hormona producida usualmente por el hígado y los riñones. Cada megacariocito produce entre 5.000 y 10.000 plaquetas. Las plaquetas son destruidas por fagocitosis en el bazo y por las células de Kupffer en el hígado. Una reserva de plaquetas es almacenada en el bazo y son liberadas cuando se necesitan por medio de contracción esplénica mediada por el sistema nervioso simpático. Formación de trombos
La función plaquetaria consiste en el mantenimiento de la hemostasia; Esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio.
Activación
La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP. Las células endoteliles producen una proteína llamada factor de von Willebrand (FvW),un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas. Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo. Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio. La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria; estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
Las plaquetas activadas cambian su forma haciéndose más esféricas, y formando pseudopodos en su superficie. De esta forma toman una forma estrellada.
Secreción de gránulos
Las plaquetas contienen gránulos alfa y gránulos densos. Las plaquetas activadas excretan el contenido de estos gránulos dentro de sus sistemas canaliculares y en la sangre circundante. Existen dos tipos de gránulos:
Gránulos densos (contienen ADP o ATP, calcio, y serotonina) Granulos-α (contienen factor 4 plaquetario, factor de crecimiento transformante beta 1 (TGF beta 1), factor de crecimiento derivado de plaquetas, fibronectina, B-tromboglobulina, FvW, fibrinógeno, y factores de coagulación factor V y XIII). Síntesis de tromboxano A2
La activación plaquetaria inicia la vía del ácido araquidónico para producir Tromboxano A2; el tromboxano A2 está involucrado en la activación de otras plaquetas y su formación es inhibida por los inhibidores de la COX, como el ácido acetilsalicílico. [editar] Adhesión y agregación La agregación plaquetaria, usa el fibrinógeno y el FvW como agentes conectores. El receptor de agregación plaquetaria más abundante es la glicoproteina IIb/IIIa (gpIIb/IIIa); se trata de un receptor para el fibrinógeno dependiente del calcio, fibronectina, vitronectina, trombospondina, y factor de von Willebrand (FvW). Otros receptores incluyen el complejo GPIb-V-IX (FvW) y GPVI (colágeno).ó Las plaquetas activadas se adherirán, via glicoproteína (GP) Ia, al colágeno expuesto por el daño epitelial. La agregación y adhesión actúan juntos para formar el tapón plaquetario. Los filamentos de Miosina y actina en las plaquetas son estimuladas para contraerse durante la agregación, reforzando todavía más el tapón. La agregación plaquetaria es estimulada por el ADP, tromboxano, y la activación del receptor-α2, pero inhibido por agentes antiinflamatorios como las prostaglandinas PGI2 y PGD2. La agregación plaquetaria se ve aumentada por la administración exógena de esteroídes anabólicos.
Reparación de heridas
El coágulo sanguíneo es solo una solución temporal para detener la hemorragia; la reparación del vaso debe ocurrir después. La agregación plaquetaria ayuda en este proceso mediante la secreción de sustancias químicas que promueven la invasión de fibroblastos del tejido conectivo adyacente hacia el interior de la herida para formar una costra. El coágulo obturador es lentamente disuelto por la enzima fibrinolítica, plasmina, y las plaquetas son eliminadas por fagocitosis. Otras funciones
Retracción del coágulo Pro-coagulación Inflamación Señalización citoquínica Fagocitosis Señalización citoquínica
Adicionalmente a su función de ser el efector celular de la hemostasia, las plaquetas son rápidamente depositadas en sitios de lesión o infección, y potencialmente modulan los procesos inflamatorios por medio de la interacción con leucocitos y por la secreción de citoquinas, quimiosinas, y otros mediadores de la inflamación. Las plaquetas también secretan factor de crecimiento derivado de plaquetas (PDGF).
El recuento de plaquetas de un individuo sano se encuentra entre 150,000 y 450,000 por μl (microlitro) de sangre (150-450 x 109/L).18 El 95 % de los individuos sanos tendrán recuentos de plaquetas dentro de este rango. Algunos tendrán recuentos de plaquetas estadísticamente anormales sin tener ninguna anormalidad demostrable. Sin embargo, si el recuento es muy alto o muy bajo la probabilidad de que una anormalidad este presente es más alta. Tanto la trombocitopenia como la trombocitosis pueden manifestarse como problemas de coagulación. En general, los recuentos bajos de plaquetas incrementan el riesgo de sangrado; sin embargo existen excepciones. Por ejemplo la trombocitopenia inmune inducida por heparina. En la trombocitosis se puede producir trombosis, sin embargo esto sucede principalmente cuando el recuento elevado es debido a desordenes mieloproliferativos. Los recuentos de plaquetas en general, no son corregidos con transfusión a menos que el paciente esté sangrando o el recuento haya caído por debajo 5 x 109/L. La transfusión esta contraindicada en la púrpura trombocitopénica idiopática (PTI), puesto que estimula la coagulopatía. En los pacientes sometidos a cirugía, niveles inferiores a 50 x 109/L están asociados a sangrado quirúrgico anormal, y procedimientos anestésicos regionales como la anestesia epidural son evitados para niveles inferiores a 80-100 x 109/L. El recuento normal de plaquetas no es garantía de función adecuada. En algunos estados, las plaquetas, siendo normales en número, son disfuncionales. Por ejemplo, el ácido acetilsalicílico interrumpe irreversiblemente la función plaquetaria mediante la inhibición de la ciclooxigenasa-1 (COX1), y por consiguiente la hemostasia normal. Las plaquetas resultantes no tienen ADN y son incapaces de producir nueva ciclooxigenasa. La función plaquetaria normal no se restaurara hasta que el uso de ASA haya cesado y un número suficiente de las plaquetas afectadas hayan sido reemplazadas por nuevas, lo cual suele tardar unos 7 días. El Ibuprofeno, un AINE, no tiene un período tan largo de efecto, y la función plaquetaria vuelve a la normalidad dentro de las 24 horas,19 y tomando ibuprofeno antes que el ASA prevendrá los efectos irreversibles de esta.20 La Uremia, a consecuencia de la insuficiencia renal, conduce a la disfunción plaquetaria que puede ser aminorada con la administración de desmopresina.
youel diaz 86760 La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón. La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca: En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA. En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno.
El interior de los vasos sanguíneos está revestido por una capa de células endoteliales las cuales actúan inhibiendo la activación plaquetaria mediante la producción de ciertos factores. Además producen una proteína llamada factor de von Willebrand (vWF), el cual permite a las células endoteliales a adherir el colágeno a la membrana basal. Cuando la capa endotelial es lesionada, el colágeno, el vWF y el factor tisular del endotelio son expuestos al flujo sanguíneo. Las plaquetas hacen contacto con el colágeno o el vWF son activadas. La activación plaquetaria resulta en el transporte de fosfolípidos cargados a la superficie plaquetaria que proporcionan una superficie para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación. Las plaquetas se hacen más esféricas y forman pseudopodos en su superficie. Estas contienen gránulos alfa y gránulos densos, excretan el contenido de estos gránulos dentro de sus sistemas canaliculares y en la sangre circundante. Existen dos tipos de gránulos: Gránulos densos (contienen ADP o ATP, calcio, y serotonina) y Granulos-α (contienen factor 4 plaquetario, factor de crecimiento transformante beta 1 (TGF beta 1), factor de crecimiento derivado de plaquetas, fibronectina, B-tromboglobulina, FvW, fibrinógeno, y factores de coagulación factor V y XIII).
1. La coagulación comienza en respuesta a una lesión en un vaso sanguíneo. Es un proceso donde se producen una serie de reacciones en cadena de varios tipos celulares y proteínas para formar un coágulo evitando asi la pérdida excesiva de sangre. Un coágulo consiste en una red de proteínas insolubles como la fibrina con plaquetas y células atrapadas que bloquea la salida de sangre hasta que se repare el tejido.
2. En la fase de iniciación es fundamental la formación de pequeñas cantidades de trombina. La lesión expone las proteínas solubles de la sangre a tejidos circundantes y a la matriz extracelular. Estos genera una serie de reacciones que tiene lugar cuando el factor VIIa se une a los factores tisulares en las membranas de las células que están fuera de los vasos originando el complejo VIIa/TF. Activa a su vez pequeñas cantidades de factor IX y X. El factor Xa se asocia con el factor Va en la superficie de las células que presentan los factores tisulares.
3. Se forma un complejo protrombinasa que da origen a pequeñas cantidades de trombina (factor IIa). El factor V puede ser activado por factor Xa, por proteasas de la matriz extracelular o ser liberado de las plaquetas al contactar con el colágeno u otros componentes de la matriz extracelular.
En la fase de amplificación se produce la activación de las plaquetas por la trombina generada en la fase de iniciación. Factores en la superficie de las plaquetas provocan la producción masiva de trombina. Tiene lugar sólo si la lesión permite la salida de plaquetas y del complejo VIII/vWF. Se secretan formas parcialmente activadas de factor V en la superficie. Se disocia el complejo VIII/vWF, permitiendo al factor vWF mediar la adhesión y agregación adicional de plaquetas y al factor VIII activarse y unirse a la membrana de las plaquetas. La trombina activa el factor XI en la superficie de la plaqueta durante esta fase.
En la fase de propagación se produce trombina de forma masiva para la formación de un coágulo estable. El factor IXa, generado en la fase de iniciación, se une al factor VIIIa en la superficie de las plaquetas. El factor XIa en la superficie de plaquetas permite la unión de más cantidad de factor IXa. El factor Xa generado por el complejo IXa/VIIIa formado en la membrana de las plaquetas se une con el factor Va. Al ensamblarse la protrombinasa (Xa/Va) provoca una masiva producción de trombina en cantidad suficiente para que se forme el coágulo.
4. Via Intrinseca: requiere de los factores de coagulación VIII, IX, X, XI y XII y de proteínas como la precalicreina (PK) y el quininógeno de alto peso molecular (HK o HMWK), al igual que iones de calcio y fosfolípidos secretados por las plaquetas. Los componentes resultan en la conversión del factor X al factor Xa. La iniciación de la vía intrínseca ocurre cuando la precalicreina, HMWK, factor XI y el factor XII son expuestos a una superficie de carga negativa, puede ocurrir como el resultado de una interacción con los fosfolípidos de las partículas lipoproteínas circulantes tales como los quilomicrones, VLDL y LDL oxidada.
5. Via Extrinseca: iniciada en el sitio de la lesión en respuesta a la liberación del factor tisular. Este es un cofactor en la activación catalizada del factor X por el factor VIIa, una proteasa de serina que contiene un residuo gla, permite transformación del factor X en factor Xa.
Los coágulos sanguíneos se presentan cuando la sangre se endurece pasando de líquida a sólida. Un coagulo que se forma dentro de una de las venas o las arterias y corazón se denomina trombo. Un trombo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo. Los trombos arteriales son formados por plaquetas y al microscopio de luz se observan de color blanco, por eso se llaman trombos blancos o arteriales. Los trombos venosos están formados por hilos de fibrina, que atrapan glóbulos rojos y al microscopio se observan de este color.
Fibrinolisis
Es un proceso normal que impide que los coágulos sanguíneos crezcan y causen problemas. La fibrinólisis primaria se refiere a la descomposición normal de los coágulos. La fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa provocando sangrado intenso.
Sistema Fibrinolitico
La fibrinolisis es un proceso enzimático de activadores e inhibidores que regulan la conversión del plasminógeno, en la enzima activa plasmina. La producción de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina. • Plasminógeno: una glicoproteína que se sintetiza en el hígado y su vida media de 2,2 días. Se une a la fibrina y van a regular la fibrinolisis. También se une a la célula endotelial lo que favorece su activación a nivel local. La activación del plasminógeno a plasmina tiene lugar por acción de los diferentes activadores.
• La precalicreína o el quininógeno de alto peso molecular (HMW-K) pueden inducir la activación del plasminógeno. La activación del factor XII conduce a la generación de calicreina que va a ser un potente activador de la fibrinolisis intrínseca.
• El activador tisular de plasminogeno (t-PA) se fija a la fibrina y forma complejos inactivos entre ambos por tanto, la actividad fibrinolítica plasmática puede aumentar o disminuir dependiendo de una serie de factores.
• Activadores uroquinasas actúan en la activación directa del plasminogeno.
• La activación exógena del plasminogeno se produce por la acción de diferentes sustancias exógenas administradas con fines terapéuticos.
• La Antitrombina VI produce reacciones fibrinolíticas que se desarrollan tras la formación de la fibrina.
ACTIVACIÓN PLAQUETARIA Un enorme número de sustancias puede actuar sobre las plaquetas e inducir su activación [el factor de activación plaquetaria,6 trombina, el tromboxano (TXA2), fosfato de adenosina (ADP),10 epinefrina, vasopresina y serotonina] a través de sus respectivos receptores, pero todas lo hacen a merced de rutas de señalización intracelular que involucran un sistema de proteínas G acoplado a enzimas efectoras comolas fofolipasas A2 (PLA2) y C (PLC) y adenilato y guanilato ciclasas.5, 6, 10 Hasta ahora se han identificado nueve componentes diferentes de la familia de proteínas G en las plaquetas y, dependiendo del tipo de proteína G que participe en la señalización. Se puede activar o inhibir el efecto.6 Un ejemplo lo constituye la regulación por proteína G de la adenilato ciclasa y por tanto de la producción de monofosfato de adenosina cíclico (AMPc). Un incremento de las cantidades de AMPc bloquea la liberación de calcio en el STD e inhibe la función de la plaqueta. Este es el mecanismo que utilizan las prostaciclinas del endotelio vascular sano para inhibir la activación plaquetaria.
Las plaquetas o trombocitos se encuentran en número de 150.000 a 400.000 por mm3 de sangre. Las plaquetas son los elementos formes más pequeños de la sangre. Tienen un diámetro de unas 2μ. Son corpúsculos anucleados con multitud de gránulos citoplasmáticos que son segregados durante su activación. Se forman en la médula ósea, mediante un proceso denominado trombopoyesis. En condiciones normales se forman 40.000 mm3/día.
Las funciones plaquetarias son: •Mantenimiento de la integridad vascular. •Interrupción inicial de la hemorragia, mediante la formación del tapón plaquetario, clavo plaquetario o trombo blanco. •Estabilización del tapón mediante los factores necesarios para la formación de fibrina. •Retracción del trombo. •Restauración del endotelio vascular mediante la producción de factores de crecimiento.
Trombopoyesis •De la célula precursora se diferencian los megacarioblastos, después los megacariocitos y al fragmentarse dan lugar a las plaquetas.
Keyla J Perez Gonzalez 87187 Hemostasia primaria Es el conjunto de fenómenos que lleva a la formación del tapón plaquetario, primer paso en la detención de la hemorragia, impidiendo la salida de elementos formes de la sangre. Durante esta fase intervienen dos mecanismos: uno vascular y otro plaquetario. •a) Espasmo vascular. De manera inmediata a la producción de la rotura del vaso, se produce una potente contracción de las fibras musculares del mismo. El resultado es una vasoconstricción que disminuye el calibre del vaso, e incluso si es pequeño puede llegar a cerrarse, disminuyendo la pérdida de sangre. •b) Formación del tapón plaquetario. En la formación del tapón plaquetario pueden distinguirse las siguientes etapas: 1.Adhesión o adherencia plaquetaria. 2.Secreción y agregación plaquetaria.
Adhesión o adherencia plaquetaria Tras la ruptura del endotelio vascular las plaquetas se adhieren a las estructuras subendoteliales, principalmente a las fibras de colágeno que afloran por le superficie rota y entran en contacto con las plaquetas. En este proceso las plaquetas pierden su forma discoide, haciéndose esféricas y emitiendo espículas por medio de las cuales se adhieren al tejido circundante. En el proceso de adhesión se precisan varias glucoproteínas de la membrana plaquetaria, el factor de von Willebrand plasmático y el colágeno y la membrana basal subendoteliales. Este proceso dura muy poco, unos 2-3 segundos. Secreción y agregación plaquetaria Se llama agregación al proceso por el cual las plaquetas se fijan unas a otras. Este proceso requiere Ca++ y ADP que deben liberarse de los gránulos plaquetarios mediante un proceso denominado activación o secreción plaquetaria. Las plaquetas sufren una profunda transformación estructural. Las membranas de los gránulos densos se unen con la membrana plasmática liberando su contenido al exterior y los gránulos α liberan su contenido. Las sustancias liberadas tienen muy diferentes tipos de actividad biológica: •Estimulan los cambios estrucuturales de las propias plaquetas. •Aumentan la adherencia plaquetaria y la secreción de más gránulos plaquetarios. •Aumentan el reclutamiento y activación de más plaquetas. •Favorecen la agregación y la coagulación. Esta secreción produce más modificaciones en las plaquetas adheridas y atrae a otras plaquetas, para irse agregando paulatinamente. Las plaquetas se mantienen unidas entre sí por puentes de enlace entre sus membranas y el tejido subendotelial. De esta forma se ha establecido una barrera, aún permeable por los espacios que quedan libres entre las plaquetas, pero que forma una línea de defensa inicial, el tapón plaquetario, o trombo blanco, para la posterior actuación del proceso de la coagulación.
Hemostasia secundaria o coagulación Es un proceso que modifica el estado líquido de la sangre dándola una estructura de tipo gel. Consiste en la transformación de una proteína soluble, el fibrinógeno, en una proteía insoluble: la fibrina; formando una malla o red que encierra elementos formes (coágulo), fortaleciendo así la unión entre plaquetas con el objeto de impedir de forma definitiva la hemorragia. De forma esquemática se puede representar como una cascada enzimática realizada por y sobre proteínas plasmáticas. Tiene varias fases: 1.Formación de protrombinasa o activador de protrombina. 2.Formación de trombina. 3.Formación de fibrina.
Formación de protrombinasa Puede seguir dos vías: •Vía extrínseca, extravascular o exógena (ver esquemas de la presentación en material complementario). •Vía intrínseca, intravascular o endógena (ver esquemas de la presentación en material complementario). Las dos vías coinciden activando el factor X para a partir de este punto formar la vía final común. Este factor junto con el factor plaquetario 3, el calcio y el factor V forma un complejo enzimático denominado protrombinasa o activador de la protrombina.
Formación de trombina Se realiza en una única reacción sobre la protrombina (Factor II). En la sangre se encuentra presente una proteína inactiva, el Factor I o fibrinógeno. La trombina cataliza el fraccionamiento de esta molécula formando monómeros de fibrina, solubles e inestables que en presencia de Ca++ y Factor XIII activado se polimerizan; formando un polímero insoluble en forma de red o malla tridimensional que cierra los espacios entre las plaquetas y sella de forma definitiva el tapón plaquetario, dando lugar al trombo rojo o coágulo.
Fibrinolisis o resolución tras la coagulación Esta última fase tiene lugar una vez que la pared vascular se ha reconstituido de nuevo, y ya no se requiere la presencia del coágulo. Este proceso se denomina fibrinolisis y consiste en la eliminación de la fibrina. Su importancia es mayor bajo el punto de vista de control en la prevención de la formación de coágulos, que en la eliminación de los mismos. El equilibrio entre la formación de fibrina y su eliminación contribuye a la limitación del proceso hemostático a la región circundante al punto de lesión. La reacción fundamental es la conversión de una proteína plasmática inactiva el plasminógeno en una activa la plasmina. Esta activación es realizada por factores endógenos como el factor activador del plasminógeno presente en las células endoteliales o la eritrocinasa presente en células sanguíneas.
Sistemas anticoagulantes La prevención de la coagulación sanguínea en el sistema vascular es un capítulo importante, ya que tan relevante es la formación de un coágulo como su limitación a un tamaño adecuado evitando que se produzca una coagulación indiscriminada. La superficie endotelial es uno de los mejores factores de seguridad ya que el mantenimiento de su integridad es una garantía para impedir la activación de la hemostasia. Las proteínas de membrana de la célula endotelial repelen los factores de coagulación. Una de estas proteínas es la trombomodulina que actúa como un receptor para la trombina uniéndose a ella y dando lugar a la activación de unas proteínas plasmáticas (C y S) que inactivan factores de coagulación y bloquean la formación de trombina. Los propios hilos de fibrina absorben entre el 85% y el 90% de la trombina formada, limitando su difusión y su acción proteolítica. Otros inhibidores de la trombina son la antitrombina III que se une a ella inactivándola; y la α2-macroglobulina y la α1-antitripsina. La heparina es un glucosaminoglucano secretado por los mastocitos y leucocitos basófilos que es administrado cuando se requiere una acción anticoagulante rápido. Su mecanismo de acción es unirse a la antitrombina III y potenciar su acción. Otros anticoagulantes funcionan secuestrando el calcio e impidiendo de esta forma la coagulación, como el citrato sódico, oxalato sódico o EDTA sódico. O los denominados anticoagulantes indirectos (cumarinas) que bloquean la absorción de la vitamina K impidiendo la síntesis proteica en el hígado de los factores de coagulación II, VII, IX y X.
Clarilix Rivera Franco 88559Activacion Plaquetaria Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo. Las células endoteliales producen una proteína llamada factor de von Willebrand (FvW), un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas. Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio. La activación plaquetaria produce el transporte de fosfolípidos cargados a la superficie de la plaqueta, estos fosfolípidos proporcionan una superficie catalítica. El calcio es element esencial para la activación de los factores de coagulación y forma parte de la activacion de mucho de los factores.
Coagulacion de la sangre 1 Luego de una lesion el factor de Von Willebrand se une al colágeno subendotelial. Las plaquetas se van a pegar al factor de Von Willebrand a través de la glicoproteína g1b, solo en el sitio de la lesión. Las plaquetas van a liberar ADP y Calcio necesario para la activación de la cascada de coagulacion. El ADP se pega a su receptor y hace que se exprese en la superficie de la plaquerta la glicoproteína IIb/IIIa. El firbinogeno se une a la glicoproteína IIb/IIIa y crea la agregación plaquetaria.
Coagulacion de la Sangre 2 Comenzando con la cascada intrínseca, después que se forma el trombo primario, este libera ADP que va a activar al Factor XII de coagulación o factor de Hageman. Con la ayuda de calcio el Factor XII va a activar al Factor XI o de Tromboplastina plasmática. Nuevamente calcio entra y El factor XIa produce el Factor IX o de Chrismas. El factor IXa promueve el cambio de el factor X a factor Xa. La cascada extrinsica comienza cuando el factor III tisular promueve la estabilidad del factor VII y en conjunto con el ADP promueven la activación plaquetaria. El factor Va y calcio intervienen para que el Factor Xa de convierta de protrombina a trombina o factor IIa. La trombina es la que cambia el fibrinógeno de la plaqueta por monómeros de fibrina los cuales finalmente promueven la coagulación de las plaquetas y la fibrinosis.
Coagulacion de la sangre 3 Luego de completada la cascada y la activación de la plaquetas, estas plaquetas se agregan entre si para formar un coagulo. Las plaquetas activadas, liberan sustancias químicas que atraen mas plaquetas y esto engrandece la vegetación. A esto se le suma la llegada de macrófagos que forman una capa sobre ellas
Clarilix Rivera Franco 88559 Coagulacion de la sangre 4 Comenzando con la cascada intrínseca, después que se forma el trombo primario, este libera ADP que va a activar al Factor XII de coagulación o factor de Hageman. Con la ayuda de calcio el Factor XII va a activar al Factor XI o de Tromboplastina plasmática. Nuevamente calcio entra y El factor XIa produce el Factor IX o de Chrismas. Este factor puede permanecer en circulación hasta 24 horas. Luego el factor IXa promueve el cambio de el factor X a factor Xa. Este factor puede permanecer en circulación hasta 48 horas. La cascada extrinsica comienza cuando el factor III tisular promueve la estabilidad del factor VII y en conjunto con el ADP promueven la activación plaquetaria. El factor Va y calcio intervienen para que el Factor Xa de convierta de protrombina a trombina o factor IIa. La trombina es la que cambia el fibrinógeno de la plaqueta por monómeros de fibrina los cuales finalmente promueven la coagulación de las plaquetas y la fibrinosis
Trombo y Coagulo Despues de la lesión y una vez realizada la cascada de coagulación, las plaquetas continúan agregándose. Con la llegada de los macrófagos y linfocitos como medio de respuesta inflamatoria, la vegetación queda cubierta capa esponjosa que se conoce como un trombo. Problemas como la hipertensión o cualquier aumento en el flujo de los hematíes a través de la arteria previamente lesionada, puede provocar que este trombo se desprenda, y una vez en circulación se le reconoce como embolo.
Fibrinolisis La lesión provoca la agregación de plaquetas con el fin de reparar el daño. Esto activa la cascada de coagulación como medio de respuesta inflamatoria importante para la formación de los monómeros de fibrina, que finalmente culminan por sellar la lesión. Esta lesión, aunque reparada se encuentra en mayor riesgo de sufrir rotura y provocarle al paciente un daño mayor, hemorragias, y que entre a circulación el llamado Trombo que pudiera provocar daños mayores en arterias pequeñas distales a la lesión principal.
Sistema Fibrinolitico El plasminogeno puede ser activado por diferentes enzimas como la calicreina(de la via intriseca), la estreproquina(via exógena), la uroquinasa (de la via endogena) y el activado plasminogeno tisular (via extrinsica). El plasminogeno activado promueve la activación de plamina. La plasmina libera sustancias que promueven la acción de las plaquetas como PDF(antitrombina VI) y el fibrinógeno. Pero este paso suele estar grandemente controlado para evitar la agregación plaquetaria exagerada. Una vez continua aumentado el fibrinógeno y la formación de fibrina, de manera retroalimentación se libera enzimas como la α2 antiplamina y la α2 macroglobulina que controlan la excesiva formación de coagulos.
VIVIANA FLAQUER 85421 DIAPOSITIVA #UNO(1) Activación plaquetaria Un enorme número de sustancias puede actuar sobre las plaquetas e inducir su activación [el factor de activación plaquetaria,6 trombina, el tromboxano (TXA2), fosfato de adenosina (ADP),10 epinefrina, vasopresina y serotonina] a través de sus respectivos receptores, pero todas lo hacen a merced de rutas de señalización intracelular que involucran un sistema de proteínas G acoplado a enzimas efectoras comolas fofolipasas A2 (PLA2) y C (PLC) y adenilato y guanilato ciclasas.5, 6, 10 Hasta ahora se han identificado nueve componentes diferentes de la familia de proteínas G en las plaquetas y, dependiendo del tipo de proteína G que participe en la señalización. Se puede activar o inhibir el efecto.6 Un ejemplo lo constituye la regulación por proteína G de la adenilato ciclasa y por tanto de la producción de monofosfato de adenosina cíclico (AMPc). Un incremento de las cantidades de AMPc bloquea la liberación de calcio en el STD e inhibe la función de la plaqueta. Este es el mecanismo que utilizan las prostaciclinas del endotelio vascular sano para inhibir la activación plaquetaria. DIAPOSITIVA #DOS (2) Coagulación de la Sangre Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre. Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional. El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse. Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas. Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc. En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares. A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
VIVIANA FLAQUER 85421 DIAPOSITIVA # CUATRO (4) El punto en el cual las dos vías se convergen es en la activación del factor X al factor Xa. El factor Xa activa a la protrombina (factor II) para convertirse en trombina (factor IIa). La trombina, a su vez, convierte el fibrinógeno a fibrina. La activación de la trombina ocurre en la superficie de las plaquetas activadas y requiere de la formación de un complejo de protrombinasa. Este complejo está compuesto de los fosfolípidos de las plaquetas: fosfatidilinositol y fosfatidilserina, Ca2+, factores Va y Xa y protrombina. El factor V es un cofactor en la formación del complejo de protrombinasa y tiene un papel similar al del factor VIII en la formación del complejo de tenasa. Como la activación del factor VIII, el factor V es activado al factor Va a través de cantidades diminutas y es inactivado por niveles aumentados de trombina. El factor Va se une a receptores específicos en la superficie de las plaquetas activadas y forma un complejo con la protrombina y el factor Xa. La protrombina es una proteína de cadena simple de 72,000 Daltons, que contiene diez residuos gla en la región de su N-terminal. Dentro del complejo de protrombinasa, la protrombina es clivada en 2 sitios por el factor Xa. Este clivaje genera una molécula de trombina activa de 2 cadenas que contiene una cadena A y una cadena B las cuales están unidas por una unión simple de disulfuro. Además de su papel en la activación de la formación del tampón de fibrina, la trombina juega un papel regulador importante en la coagulación. La trombina se une con la trombomodulina presente en la superficie de las células endoteliales formando un complejo que convierte la proteína C a la proteína Ca. La proteína S y la proteína Ca son cofactores que degradan a los factores Va y VIIIa y así limitando la actividad de estos 2 factores en la cascada de la coagulación. La trombina también se une, resultando en la activación de la señalización, a una clase de receptores acoplados a proteínas G llamados receptores activados por proteasas (siglas en Inglés: PARs), específicamente PAR-1, -3 y -4. Los PARs utilizan un mecanismo único para convertir el producto del clivaje proteolítico extracelular a un evento de señalización intracelular. Los PARs llevan su propio ligando el cual se mantiene inactivo hasta que el clivaje por las proteasas libere al ligando, por ejemplo como es el caso de la trombina. El ligando liberado reacciona con un dominio de unión del ligante del PAR lo que resulta en la activación de varias cascadas de señalización. Estas cascadas de señalización resultan en una liberación aumentada de interleucinas, (ILs), IL-1 e IL-6, una secreción aumentada de la molécula de adhesión intercelular-1 (ICAM-1) y la molécula de adhesión de la célula vascular-1 (VCAM-1). La señalización inducida por la trombina también resulta en un aumento en la activación plaquetaria y en la adhesión de leucocitos. La trombina también activa al inhibidor de fibrinólisis activado por la trombina (TAFI) y así modulando la fibrinólisis (degradación de los coágulos de fibrina). El TAFI también se conoce como la carboxipeptidasa U (CPU) cuya actividad resulta en la eliminación de las lisinas en la C-terminal de la fibrina parcialmente degradada. Esto conlleva a una falla en la activación del plasminógeno, y así reduciendo la taza de la disolución del coágulo de fibrina (esto se llama fibrinólisis).
VIVIANA FLAQUER 85421 DIAPOSITIVA #CINCO (5) *Vía Intrínseca Recibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso hemostático. El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular. En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación. En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como la celulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción. A esta vía es posible subdividirla en tres pasos: -Formación del factor XIa En esta etapa participan cuatro proteínas: Precalicreína, Quininógeno de alto peso molecular (HMWK) y los factores XII y XI. Esta etapa no requiere de iones calcio. Estos cuatro factores se adsorben sobre la superficie cargada negativamente, formando el complejo cebador o de iniciación. De estos factores el XII funciona como verdadero iniciador, ya que si bien es una proenzima, posee una pequeña actividad catalítica que alcanza para activar a la precalicreína convirtiéndola en calicreína. En segunda instancia la calicreína actúa catalíticamente sobre el factor XII para convertirlo en XIIa, una enzima muchísimo más activa. La actividad catalítica de la calicreína se ve potenciada por el HMWK. Por último la proteasa XIIa actúa sobre el factor XI para liberar XIa. -Formación del factor IXa El factor IX se encuentra en el plasma como una proenzima. En presencia de iones Ca2+ el factor XIa cataliza la ruptura de una unión peptídica en la molécula del factor IX para formar un glucopéptido de 10 KDa y liberar por otro lado al factor IXa. El factor IX se encuentra ausente en personas con hemofilia tipo B. -Formación del factor Xa Sobre la membrana de las plaquetas se forma un complejo constituido por los factores IXa, X y VIII. Los residuos gamma-carboxiglutamato de los factores IXa y X actúan como quelantes del ion Ca2+, permitiendo que estos componentes formen un complejo unido por medio de puentes de iones calcio y ayudando a que el complejo se ancle a los fosfolípidos de membrana. Primero se unen los factores X y IXa a la membrana y luego se une el VIII. El factor VIII es en realidad un homorodímero, formado por cuatro cadenas proteicas, cada una codificada por un gen diferente (VIII:C y VIII:R). El componente VIII:C es conocido como "componente antihemofílico" y actúa como cofactor del IXa en la activación del factor X, el componente VIII:R es el que permite la unión del factor VIII al complejo. La ausencia del componente antihemofílico causa hemofilia A. El complejo formado por los factores IXa-X-VIII-Fosfolípidos y Ca2+ actúa sobre el factor X para convertirlo en Xa. En este punto concluye la vía intrínseca.
VIVIANA FLAQUER 85421 CONTINUACION DIAPOSITIVA #CINCO (5) El factor Xa y la protrombina se adsorben sobre la membrana utilizando iones Ca2+ como puentes. El factor Va se une a la protrombina acelerando la reacción. El factor Va se produce por la acción de la trombina sobre el factor V en un claro ejemplo de una reacción que va acelerándose a medida que progresa (reacción autoacelerada). -Formación de fibrina El fibrinógeno (factor I) es una glicoproteína compuesta por seis cadenas polipeptídicas: dos A-alfa, dos B-beta y dos gamma; unidas entre sí por puentes disulfuro. Se trata de una molécula alargada y simétrica formada por tres dominios globulares conectados por segmentos fibrilares. Cada mitad de la molécula se encuentra formada por tres cadenas (A-alfa, B-beta y gamma) que se enrollan en una triple hélice muy compacta en los sectores fibrilares. Los extremos amino de las seis cadenas se reúnen en el dominio globular central. En un hecho que parecería muy curioso, los extremos N-terminales de las cadenas A-alfa y B-beta emergen como cabos libres del dominio globular central. Representación de la molécula de fibrinógeno y cómo, al eliminarse los fibrinopéptidos, polimeriza para formar un agregado de fibrina. Estas cadenas son muy ricas en aspartato y glutamato, además las cadenas B-beta poseeen en esta región residuos tirosina-O-sulfato formados postraduccionalmente. Estos residuos con una alta tendencia a adquirir carga negativa contribuyen a formar una región central con una muy alta densidad de carga. Esta región electronegativa central es la responsable de la repulsión entre moléculas de fibrina que las mantiene en solución. La trombina ataca los enlaces arginina-glicina presentes en estos "cabos libres", separando cuatro péptidos; dos segmentos A de 18 aminoácidos cada uno (provenientes de las cadenas A-alfa), y dos segmentos B de 20 aminoácidos (provenientes de las cadenas B-beta). A estos péptidos se los suele denominar "fibrinopéptidos". El resto que queda de la molécula es un monómero de fibrina de composición alfa2beta2gamma2. Al eliminarse los fibinopéptidos desaparecen las fuerzas de repulsión intermoleculares con lo que los monómeros de fibrina tienden a agruparse espontáneamente formando asociaciones altamente ordenadas. Los monómeros se disponen uno a continuación del otro, cabeza con cabeza en forma de largas hebras. Estas hebras a su vez forman manojos, emparejándose con otras hebras de tal manera que la región central de los monómeros de fibrina de una se encuentra rodeada por las cabezas de los monómeros de fibrina de las otras. Este emparejamiento se hace posible gracias a interaciones de tipo electrostático y puente hidrógeno entre las regiones centrales de los monómeros de una y las cabezas globulares de otras. -Entrecruzamiento de la fibrina Los haces paralelos de fibrina polimerizada forman una asociación laxa, que se encuentra en equilibrio con la forma monomérica de la molécula; por lo que sería imposible que cumplieran su papel de formar un coágulo estable sin reforzar esta estructura por medio de enlaces covalentes entre hebras vecinas. La formación de estos "puentes" covalentes intercatenarios es catalizada por la enzima transglutaminasa (conocida también como factor XIIIa). La transglutaminidasa cataliza la formación de enlaces amida entre restos glutamina y lisina de hebras próximas entre sí. En la reacción se libera amoníaco en forma de ion amonio (NH4+). Esta enzima se forma a partir del factor XIII por acción de la trombina.
DIAPOSITIVA #SEIS (6) Trombo & Cuagulo Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida. Un coágulo sanguíneo que se forma dentro de una de las venas o las arterias se denomina trombo y también se puede formar en el corazón. Un trombo que se desprende y viaja desde un lugar en el cuerpo a otro se llama émbolo.Un trombo o émbolo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo. Una obstrucción en una arteria puede impedir que el oxígeno llegue a los tejidos en esa área, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede provocar daños en los tejidos o la muerte. Una obstrucción en una vena generalmente provocará acumulación de líquido e hinchazón. Las situaciones en las que es más probable que se forme un coágulo de sangre abarcan: Estar en reposo en cama por largo tiempo. Cruzar las piernas durante largos periodos al estar sentado o sentarse durante mucho tiempo, como en un avión o en un vehículo. Durante y después del embarazo. No tener suficiente agua en el cuerpo (deshidratación). Tomar píldoras anticonceptivas u hormonas estrógeno (especialmente en las mujeres que fuman). Utilizar un catéter intravenoso por largo tiempo. Los coágulos de sangre también son más probables en personas con cáncer, cirugía o lesión recientes, obesidad y enfermedades del hígado o del riñón. Una acumulación de colesterol que estreche una arteria puede cambiar o reducir el flujo de sangre, facilitando la formación de un coágulo sanguíneo o de un trombo. Las afecciones que se transmiten de padres a hijos (hereditarias) pueden hacerlo más propenso a la formación de coágulos sanguíneos anormales. Los trastornos hereditarios que afectan la coagulación son: Trombofilia por factor V de Leiden Mutación de la protrombina G20210A Otras afecciones raras como deficiencias de antitrombina III, proteína C y S Un coágulo de sangre puede bloquear una arteria o una vena en el corazón, afectando: El corazón (angina de pecho o un ataque cardíaco) Los intestinos (isquemia mesentérica) o trombosis venosa mesentérica Los riñones (trombosis de la vena renal) Las arterias de las piernas o brazos Las piernas (trombosis venosa profunda) Los pulmones (embolia pulmonar) El cuello o el cerebro (accidente cerebrovascular) Los trombos y émbolos pueden fijarse firmemente a un vaso sanguíneo y pueden bloquear parcial o completamente el flujo de sangre a dicho vaso. Un bloqueo en el vaso sanguíneo impide que el flujo normal de sangre y oxígeno llegue a los tejidos en ese sitio, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede ocasionar daño tisular o necrosis en esa área.
VIVIANA FLAQUER 85421 DIAPOSITIVA #SIETE (7) Fibrinólisis Después de que el coágulo se ha establecido, comienza la reparación de los tejidos afectados con el proceso de cicatrización. Para hacer posible esto el coágulo es colonizado por células que formarán nuevos tejidos y en el proceso va siendo degradado. La degradación de la fibrina (fibrinólisis), componente mayoritaria del coágulo, es catalizada por la enzima plasmina, una serina proteasa que ataca las uniones peptídicas en la región triple hélice de los monómeros de fibrina. La plasmina se genera a partir del plasminógeno, un precursor inactivo; activándose tanto por la acción de factores intrínsecos (propios de la cascada de coagulación) como extrínsecos, el más importante de los cuales es producido por el endotelio vascular. Se le denomina "activador tisular del plasminógeno" (t-PA). El gen de este factor ha sido clonado y actualmente se puede obtener la proteína producida por tecnología de ADN recombinante. Este factor suele utilizarse en clínica para favorecer la disolución de trombos. DIAPOSITIVA #OCHO (8) Sistema Fibrinolitico La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón. La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca: En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA. En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Las plaquetas son células producidas por los megacariocitos en la médula ósea mediante el proceso de fragmentación citoplasmática, circulan por la sangre y tiene un papel muy importante en la coagulación. Para ello forman nudos en la red de fibrina, liberan substancias importantes para acelerar la coagulación y aumentan la retracción del coágulo sanguíneo. En las heridas las plaquetas aceleran la coagulación, y además al aglutinarse obstruyen pequeños vasos, y engendran substancias que los contraen.
Procedimiento de obtención: Para realizar este análisis no se precisa estar en ayunas. Se puede realizar la toma en un lugar apropiado (consulta, clínica, hospital) pero en ocasiones se realiza en el propio domicilio del paciente. Para realizar la toma se precisa de localizar una vena apropiada y en general se utilizan las venas situadas en la flexura del codo. La persona encargada de tomar la muestra utilizará guantes sanitarios, una aguja (con una jeringa o tubo de extracción). Le pondrá un tortor (cinta de goma-látex) en el brazo para que las venas retengan más sangre y aparezcan más visibles y accesibles. Limpiará la zona del pinchazo con un antiséptico y mediante una palpación localizará la vena apropiada y accederá a ella con la aguja. Le soltarán el tortor. Cuando la sangre fluya por la aguja el sanitario realizará una aspiración (mediante la jeringa o mediante la aplicación de un tubo con vacío). Si se requiere varias muestras para diferentes tipos de análisis se le extraerá más o menos sangre o se aplicarán diferentes tubos de vacío. Al terminar la toma, se extrae la aguja y se presiona la zona con una torunda de algodón o similar para favorecer la coagulación y se le indicará que flexione el brazo y mantenga la zona presionada con un esparadrapo durante unas horas.
Problemas y posibles riesgos: La obtención mediante un pinchazo de la vena puede producir cierto dolor. La posible dificultad en encontrar la vena apropiada puede dar lugar a varios pinchazos. Aparición de un hematoma (moratón o cardenal) en la zona de extracción, suele deberse a que la vena no se ha cerrado bien tras la presión posterior y ha seguido saliendo sangre produciendo este problema. Puede aplicarse una pomada tipo Hirudoid® o Trombocid® en la zona. Inflamación de la vena (flebitis), a veces la vena se ve alterada, bien sea por una causa meramente física o por que se ha infectado. Se deberá mantener la zona relajada unos días y se puede aplicar una pomada tipo Hirudoid® o Trombocid® en la zona. Si el problema persiste o aparece fiebre deberá consultarlo con su médico.
Valores normales: Las alteraciones en el número de plaquetas así como en su tamaño pueden ser clave del diagnóstico. Hay una gran variación en el rango normal del recuento de plaquetas. Valores normales: De 150.000 a 400.000/mm3
Significado de los resultados anormales: La disminución en el número de plaquetas (por debajo del límite menor normal) se denomina trombocitopenia y el aumento en el número de las mismas (superior al límite normal más alto) se llama trombocitosis. Cuando existe una trombocitopenia aislada, la causa más común es la destrucción inmune, pero existen trombocitopenias asociadas a un gran número de otras enfermedades como son:
Coagulación intravascular diseminada (C.IV.D) Anemia hemolítica microangiopática Hiperesplenismo (exceso de función del bazo) Disminución de la producción en el caso de anemia aplástica, Invasión de la médula ósea por enfermedades malignas como leucemias, neuroblastoma, linfoma. Quimioterapia por cáncer Púrpura trombocitopénica idiopática (PTI) Leucemia Prótesis de válvula coronaria Transfusión de sangre Choque anafiláctico Algunas infecciones que producen hemorragias (púrpuras con trombocitopenia), en las que se hallan muy disminuidas.
La trombocitosis es el aumento en el recuento de plaquetas y puede ser secundario Las infecciones suelen ser la causa más frecuente (virales, bacterianas o por micoplasma), pero existen muchas otras enfermedades que se asocian a trombocitosis como son: Anemia por déficit de hierro Enfermedad de Kawasaki Síndrome nefrótico Síndrome post-esplenectomía (tras extraer el bazo) Traumatismos Tumores Trombocitosis primaria
La coagulación es el proceso por el cual se forma un coágulo sanguíneo. Comienza en respuesta a una lesión en un vaso sanguíneo. En el proceso de coagulación se producen una serie de reacciones en cadena en las que participan varios tipos celulares y proteínas solubles de la sangre con el objetivo de formar un coágulo para evitar la pérdida excesiva de sangre. Un coágulo consiste en una red de proteínas insolubles como la fibrina con plaquetas y células atrapadas que bloquea la salida de sangre hasta que se repare el tejido. Se distinguen dos rutas de activación de la cascada de coagulación conocidas como vía extrínseca y vía intrínseca. Hacen referencia al lugar donde se inicia la cascada de coagulación: el interior de un vaso sanguíneo (intrínseca) o fuera de un vaso sanguíneo (extrínseca). Ambas vías convergen en la activación del factor Xa que transforma la protrombina en trombina. En el paso siguiente la trombina genera fibrina a partir de fibrinógeno.
En el proceso de coagulación participan los siguientes elementos: Células que presentan el factor tisular (TF): Estas células no están normalmente en contacto con elementos solubles de la sangre. Cuando se produce una lesión con pérdida de sangre, el reconocimiento entre el TF y las proteínas solubles dispara la cascada de la coagulación. Plaquetas: Son fragmentos celulares derivados del megacariocito. Participan en el proceso de coagulación en las fases de amplificación y propagación y forman parte del coágulo. En sus membranas se dan muchas de las reacciones del proceso de coagulación y secretan varios elementos imprescindibles durante el proceso. Factores de coagulación: Son un conjunto de proteínas que participan en las reacciones enzimáticas encadenadas que hacen posible la cascada de la coagulación. Las proteínas de la cascada se van activando por proteolisis adquiriendo actividad proteasa y activando por proteolisis a la siguiente proteína de la cascada de coagulación. La activación de esta cascada lleva a la producción de grandes cantidades de trombina que posibilitan la formación del coágulo. Algunos de estos factores se asocian entre sí en la membrana de plaquetas y de otros tipos celulares que participan en el proceso de coagulación Sistema anticoagulante: Para evitar la formación de trombos (coágulos fijos dentro de los vasos) o émbolos (coágulos móviles que viajan por el torrente sanguíneo) existen una serie de proteínas que confinan la formación del coágulo a la zona lesionada. Algunos elementos de estos sistemas son la proteína C, la proteína S, la antitrombina (AT) y la trombomodulina (TM). El proceso de coagulación se puede esquematizar en las siguientes fases: Fase de iniciación. En esta fase es fundamental la formación de pequeñas cantidades de trombina. La lesión de un vaso sanguíneo expone las proteínas solubles de la sangre a las células de los tejidos circundantes y a la matriz extracelular. Estos contactos van a generar una serie de reacciones que constituyen la fase de iniciación. La cascada de reacciones que tiene lugar en esta fase comienza cuando el factor VIIa (factor VII activo) se une a los factores tisulares (TF) en las membranas de las células que están fuera de los vasos originando el complejo VIIa/TF. Este complejo activa a su vez pequeñas cantidades de factor IX y X. El factor Xa (factor X activo) se asocia con el factor Va (factor V activo) en la superficie de las células que presentan los factores tisulares. Se forma un complejo protrombinasa que origina pequeñas cantidades de trombina (factor IIa). El factor V puede ser activado por el propio factor Xa, por ciertas proteasas de la matriz extracelular o puede ser liberado directamente por parte de las plaquetas al contactar con el colágeno u otros componentes de la matriz extracelular.
Normalmente el factor Xa permanece unido a la membrana de las células que presentan factores tisulares para protegerse de la degradación. En caso de producirse su disociación y pasar al torrente sanguíneo es rápidamente inhibido en la fase fluida por inhibidores de la ruta de los factores tisulares o por antitrombina evitando que sea activo fuera de la zona de la lesión. De esta forma, la presencia de inhibidores focaliza y limita eficientemente la actividad del factor Xa únicamente a las superficies celulares donde se produce la lesión. El factor IXa puede moverse desde la célula en que se origina a través de la fase fluida hacia una plaqueta vecina u otra superficie celular ya que es inhibido mucho más lentamente por antitrombina. Fase de amplificación. En esta fase se produce la activación de las plaquetas gracias a la pequeña cantidad de trombina generada en la fase de iniciación. En esta fase factores en la superficie de las plaquetas provocan la producción masiva de trombina. La fase de amplificación tiene lugar sólo si la lesión permite la salida de plaquetas y del complejo VIII/vWF (factor von Willebrand). Durante la activación de las plaquetas se secretan formas parcialmente activadas de factor V en la superficie. En esta fase también se disocia el complejo VIII/vWF, permitiendo al factor vWF mediar la adhesión y agregación adicional de plaquetas en el sitio de la lesión y al factor VIII activarse y unirse a la membrana de las plaquetas. La trombina generada también activa el factor XI en la superficie de la plaqueta durante esta fase.
Fase de propagación. En esta fase se produce trombina de forma masiva para la formación de un coágulo estable. También acuden gran número de plaquetas al lugar de la lesión. En esta fase sólo participan las plaquetas activadas. El factor IXa, generado en la fase de iniciación, se une al factor VIIIa en la superficie de las plaquetas. El factor XIa en la superficie de plaquetas permite la unión de más cantidad de factor IXa. El factor Xa generado por el complejo IXa/VIIIa formado en la membrana de las plaquetas se asocia rápidamente con el factor Va expresado por las plaquetas en la fase de amplificación. Al ensamblarse la protrombinasa (Xa/Va) provoca una masiva producción de trombina en cantidad suficiente para que se forme el coágulo.
Una vez formado el coágulo de fibrina y plaquetas en la zona lesionada, el proceso ha de ser controlado para evitar la oclusión trombótica en las zonas no dañadas. La proteína C, la proteína S y la trombomodulina constituyen un importante sistema de control que restringe la coagulación a la zona de la lesión. Durante el proceso de coagulación parte de la trombina formada puede difundir por los vasos sanguíneos. Cuando llega a una célula endotelial intacta se une a la trombomodulina en la superficie endotelial. El complejo trombomodulina/trombina activa a la proteína C que se une a la proteína S e inactiva los factores Va y VIIIa. De esta forma se detiene la generación de trombina en las zonas intactas. La proteína C y la proteína S son mucho más eficientes inactivando al factor Va en la superficie de las células endoteliales que en la de las plaquetas. Otro sistema para prevenir la generación de trombina en células endoteliales de zonas no dañadas es la acción de los inhibidores de proteasa antitrombina e inhibidores de la ruta de los factores tisulares (TFPI: Tissue Factor Pathway Inhibitor) que ayudan a confinar la formación de trombina a las áreas alrededor de la lesión. Los inhibidores de proteasa AT-III y TFPI pueden inhibir rápidamente proteasas generadas cerca de un endotelio intacto.
Existen diversas enfermedades derivadas de fallos en algunos de estos elementos que participan en el proceso de coagulación. Pueden tener un origen genético o no y ser o no reversibles. Existen tres grupos de genes asociados a defectos en la coagulación: Genes responsables de la adhesión, activación y agregación plaquetaria. Genes de biosíntesis de factores y cofactores de coagulación sanguínea. Genes involucrados en el sistema de anticoagulación y fibrinolisis. La hemofilia es un ejemplo de enfermedad causada por un defecto genético en un componente del proceso de coagulación (factor VIII en la hemofilia A y factor IX en la hemofilia B).
Los fármacos anticoagulantes que se han empleado tradicionalmente tienen sus limitaciones. Actualmente se están desarrollando nuevos anticoagulantes inhibidores del factor Xa e inhibidores directos de la trombina. Varios de ellos aún están en fase de ensayo clínico para prevención y tratamiento de la trombosis: "Dabigatran Etexilate" es un fármaco oral que se convierte en "dabigatran", un inhibidor de la trombina. La eficacia y seguridad de esta sustancia ha sido evaluada en la fase II del ensayo clínico y se encuentra en la fase III de prevención y tratamiento de la enfermedad tromboembólica venosa (VTE: Venous Thromboembolism Events) y prevención de apoplejías derivadas de fibrilación auricular. Otra diana terapéutica para el diseño de drogas anticoagulantes es el factor Xa. "Fondaparinux", es un pentasacárido obtenido por síntesis química que se une de forma rápida y específica a antitrombina en la sangre, induciendo un cambio conformacional en la antitrombina. Este cambio conformacional incrementa su afinidad por el factor Xa potenciando su función inhibidora unas 300 veces. Además cada molécula de fármaco puede modificar a varias moléculas de antitrombina. Esta sustancia ha sido probada en numerosos ensayos clínicos de fase III con diferentes dosis, obteniéndose una reducción de un 50% en la incidencia de enfermedad tromboembólica venosa.
La fibrinólisis primaria se refiere a la descomposición normal de los coágulos, mientras que la fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa.
Cuando se produce demasiado rápidamente, puede provocar hemorragias espectaculares (fibrinólisis hemorrágica) intra o postoperatorias (particularmente en Cirugía Torácica), o después de un parto, de un aborto o de un choque traumático; o bien puede provocar hemorragias menos importantes (hematuria) en el curso de ciertos cánceres (próstata, páncreas, estómago), de las cirrosis y de las leucosis. Obedece a la liberación de activadores de la profibrinolisina, o bien a la producción de un fermento proteolítico por los tejidos enfermos.
Puede ser primitiva aunque por lo general es secundaria, reactiva, en el curso de un síndrome de coagulación intravascular diseminada.
Los coágulos de sangre se forman a partir de una proteína llamada fibrina. La descomposición de la fibrina (fibrinólisis) puede incrementarse bajo ciertas condiciones, tales como: Infecciones bacterianas Ejercicio intenso Glucemia baja Oxigenación insuficiente a los tejidos
ENMANUEL REYES S. #87419 DIAPOSITIVA #1 ACTIVACIÓN PLAQUETARIA.
Un enorme número de sustancias puede actuar sobre las plaquetas e inducir su activación [el factor de activación plaquetaria,6 trombina, el tromboxano (TXA2), fosfato de adenosina (ADP),10 epinefrina, vasopresina y serotonina] a través de sus respectivos receptores, pero todas lo hacen a merced de rutas de señalización intracelular que involucran un sistema de proteínas G acoplado a enzimas efectoras comolas fofolipasas A2 (PLA2) y C (PLC) y adenilato y guanilato ciclasas.5, 6, 10 Hasta ahora se han identificado nueve componentes diferentes de la familia de proteínas G en las plaquetas y, dependiendo del tipo de proteína G que participe en la señalización. Se puede activar o inhibir el efecto.6 Un ejemplo lo constituye la regulación por proteína G de la adenilato ciclasa y por tanto de la producción de monofosfato de adenosina cíclico (AMPc). Un incremento de las cantidades de AMPc bloquea la liberación de calcio en el STD e inhibe la función de la plaqueta. Este es el mecanismo que utilizan las prostaciclinas del endotelio vascular sano para inhibir la activación plaquetaria.
DIAPOSITIVA #2 COAGULACIÓN DE LA SANGRE.
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre. Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional. El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse. Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas. Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc. En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares. A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
El punto en el cual las dos vías se convergen es en la activación del factor X al factor Xa. El factor Xa activa a la protrombina (factor II) para convertirse en trombina (factor IIa). La trombina, a su vez, convierte el fibrinógeno a fibrina. La activación de la trombina ocurre en la superficie de las plaquetas activadas y requiere de la formación de un complejo de protrombinasa. Este complejo está compuesto de los fosfolípidos de las plaquetas: fosfatidilinositol y fosfatidilserina, Ca2+, factores Va y Xa y protrombina. El factor V es un cofactor en la formación del complejo de protrombinasa y tiene un papel similar al del factor VIII en la formación del complejo de tenasa. Como la activación del factor VIII, el factor V es activado al factor Va a través de cantidades diminutas y es inactivado por niveles aumentados de trombina. El factor Va se une a receptores específicos en la superficie de las plaquetas activadas y forma un complejo con la protrombina y el factor Xa. La protrombina es una proteína de cadena simple de 72,000 Daltons, que contiene diez residuos gla en la región de su N-terminal. Dentro del complejo de protrombinasa, la protrombina es clivada en 2 sitios por el factor Xa. Este clivaje genera una molécula de trombina activa de 2 cadenas que contiene una cadena A y una cadena B las cuales están unidas por una unión simple de disulfuro. Además de su papel en la activación de la formación del tampón de fibrina, la trombina juega un papel regulador importante en la coagulación. La trombina se une con la trombomodulina presente en la superficie de las células endoteliales formando un complejo que convierte la proteína C a la proteína Ca. La proteína S y la proteína Ca son cofactores que degradan a los factores Va y VIIIa y así limitando la actividad de estos 2 factores en la cascada de la coagulación. La trombina también se une, resultando en la activación de la señalización, a una clase de receptores acoplados a proteínas G llamados receptores activados por proteasas (siglas en Inglés: PARs), específicamente PAR-1, -3 y -4. Los PARs utilizan un mecanismo único para convertir el producto del clivaje proteolítico extracelular a un evento de señalización intracelular. Los PARs llevan su propio ligando el cual se mantiene inactivo hasta que el clivaje por las proteasas libere al ligando, por ejemplo como es el caso de la trombina. El ligando liberado reacciona con un dominio de unión del ligante del PAR lo que resulta en la activación de varias cascadas de señalización. Estas cascadas de señalización resultan en una liberación aumentada de interleucinas, (ILs), IL-1 e IL-6, una secreción aumentada de la molécula de adhesión intercelular-1 (ICAM-1) y la molécula de adhesión de la célula vascular-1 (VCAM-1). La señalización inducida por la trombina también resulta en un aumento en la activación plaquetaria y en la adhesión de leucocitos. La trombina también activa al inhibidor de fibrinólisis activado por la trombina (TAFI) y así modulando la fibrinólisis (degradación de los coágulos de fibrina). El TAFI también se conoce como la carboxipeptidasa U (CPU) cuya actividad resulta en la eliminación de las lisinas en la C-terminal de la fibrina parcialmente degradada. Esto conlleva a una falla en la activación del plasminógeno, y así reduciendo la taza de la disolución del coágulo de fibrina (esto se llama fibrinólisis).
*Vía Intrínseca Recibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso hemostático. El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular. En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación. En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como la celulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción. A esta vía es posible subdividirla en tres pasos: -Formación del factor XIa En esta etapa participan cuatro proteínas: Precalicreína, Quininógeno de alto peso molecular (HMWK) y los factores XII y XI. Esta etapa no requiere de iones calcio. Estos cuatro factores se adsorben sobre la superficie cargada negativamente, formando el complejo cebador o de iniciación. De estos factores el XII funciona como verdadero iniciador, ya que si bien es una proenzima, posee una pequeña actividad catalítica que alcanza para activar a la precalicreína convirtiéndola en calicreína. En segunda instancia la calicreína actúa catalíticamente sobre el factor XII para convertirlo en XIIa, una enzima muchísimo más activa. La actividad catalítica de la calicreína se ve potenciada por el HMWK. Por último la proteasa XIIa actúa sobre el factor XI para liberar XIa. -Formación del factor IXa El factor IX se encuentra en el plasma como una proenzima. En presencia de iones Ca2+ el factor XIa cataliza la ruptura de una unión peptídica en la molécula del factor IX para formar un glucopéptido de 10 KDa y liberar por otro lado al factor IXa. El factor IX se encuentra ausente en personas con hemofilia tipo B. -Formación del factor Xa Sobre la membrana de las plaquetas se forma un complejo constituido por los factores IXa, X y VIII. Los residuos gamma-carboxiglutamato de los factores IXa y X actúan como quelantes del ion Ca2+, permitiendo que estos componentes formen un complejo unido por medio de puentes de iones calcio y ayudando a que el complejo se ancle a los fosfolípidos de membrana. Primero se unen los factores X y IXa a la membrana y luego se une el VIII. El factor VIII es en realidad un homorodímero, formado por cuatro cadenas proteicas, cada una codificada por un gen diferente (VIII:C y VIII:R). El componente VIII:C es conocido como "componente antihemofílico" y actúa como cofactor del IXa en la activación del factor X, el componente VIII:R es el que permite la unión del factor VIII al complejo. La ausencia del componente antihemofílico causa hemofilia A. El complejo formado por los factores IXa-X-VIII-Fosfolípidos y Ca2+ actúa sobre el factor X para convertirlo en Xa. En este punto concluye la vía intrínseca.
El factor Xa y la protrombina se adsorben sobre la membrana utilizando iones Ca2+ como puentes. El factor Va se une a la protrombina acelerando la reacción. El factor Va se produce por la acción de la trombina sobre el factor V en un claro ejemplo de una reacción que va acelerándose a medida que progresa (reacción autoacelerada). -Formación de fibrina El fibrinógeno (factor I) es una glicoproteína compuesta por seis cadenas polipeptídicas: dos A-alfa, dos B-beta y dos gamma; unidas entre sí por puentes disulfuro. Se trata de una molécula alargada y simétrica formada por tres dominios globulares conectados por segmentos fibrilares. Cada mitad de la molécula se encuentra formada por tres cadenas (A-alfa, B-beta y gamma) que se enrollan en una triple hélice muy compacta en los sectores fibrilares. Los extremos amino de las seis cadenas se reúnen en el dominio globular central. En un hecho que parecería muy curioso, los extremos N-terminales de las cadenas A-alfa y B-beta emergen como cabos libres del dominio globular central. Representación de la molécula de fibrinógeno y cómo, al eliminarse los fibrinopéptidos, polimeriza para formar un agregado de fibrina. Estas cadenas son muy ricas en aspartato y glutamato, además las cadenas B-beta poseeen en esta región residuos tirosina-O-sulfato formados postraduccionalmente. Estos residuos con una alta tendencia a adquirir carga negativa contribuyen a formar una región central con una muy alta densidad de carga. Esta región electronegativa central es la responsable de la repulsión entre moléculas de fibrina que las mantiene en solución. La trombina ataca los enlaces arginina-glicina presentes en estos "cabos libres", separando cuatro péptidos; dos segmentos A de 18 aminoácidos cada uno (provenientes de las cadenas A-alfa), y dos segmentos B de 20 aminoácidos (provenientes de las cadenas B-beta). A estos péptidos se los suele denominar "fibrinopéptidos". El resto que queda de la molécula es un monómero de fibrina de composición alfa2beta2gamma2. Al eliminarse los fibinopéptidos desaparecen las fuerzas de repulsión intermoleculares con lo que los monómeros de fibrina tienden a agruparse espontáneamente formando asociaciones altamente ordenadas. Los monómeros se disponen uno a continuación del otro, cabeza con cabeza en forma de largas hebras. Estas hebras a su vez forman manojos, emparejándose con otras hebras de tal manera que la región central de los monómeros de fibrina de una se encuentra rodeada por las cabezas de los monómeros de fibrina de las otras. Este emparejamiento se hace posible gracias a interaciones de tipo electrostático y puente hidrógeno entre las regiones centrales de los monómeros de una y las cabezas globulares de otras. -Entrecruzamiento de la fibrina Los haces paralelos de fibrina polimerizada forman una asociación laxa, que se encuentra en equilibrio con la forma monomérica de la molécula; por lo que sería imposible que cumplieran su papel de formar un coágulo estable sin reforzar esta estructura por medio de enlaces covalentes entre hebras vecinas. La formación de estos "puentes" covalentes intercatenarios es catalizada por la enzima transglutaminasa (conocida también como factor XIIIa). La transglutaminidasa cataliza la formación de enlaces amida entre restos glutamina y lisina de hebras próximas entre sí. En la reacción se libera amoníaco en forma de ion amonio (NH4+). Esta enzima se forma a partir del factor XIII por acción de la trombina.
ENMANUEL REYES S. #87419 DIAPOSITIVA #6 TROMBO & CUAGULO Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida. Un coágulo sanguíneo que se forma dentro de una de las venas o las arterias se denomina trombo y también se puede formar en el corazón. Un trombo que se desprende y viaja desde un lugar en el cuerpo a otro se llama émbolo.Un trombo o émbolo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo. Una obstrucción en una arteria puede impedir que el oxígeno llegue a los tejidos en esa área, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede provocar daños en los tejidos o la muerte. Una obstrucción en una vena generalmente provocará acumulación de líquido e hinchazón. Las situaciones en las que es más probable que se forme un coágulo de sangre abarcan: Estar en reposo en cama por largo tiempo. Cruzar las piernas durante largos periodos al estar sentado o sentarse durante mucho tiempo, como en un avión o en un vehículo. Durante y después del embarazo. No tener suficiente agua en el cuerpo (deshidratación). Tomar píldoras anticonceptivas u hormonas estrógeno (especialmente en las mujeres que fuman). Utilizar un catéter intravenoso por largo tiempo. Los coágulos de sangre también son más probables en personas con cáncer, cirugía o lesión recientes, obesidad y enfermedades del hígado o del riñón. Una acumulación de colesterol que estreche una arteria puede cambiar o reducir el flujo de sangre, facilitando la formación de un coágulo sanguíneo o de un trombo. Las afecciones que se transmiten de padres a hijos (hereditarias) pueden hacerlo más propenso a la formación de coágulos sanguíneos anormales. Los trastornos hereditarios que afectan la coagulación son: Trombofilia por factor V de Leiden Mutación de la protrombina G20210A Otras afecciones raras como deficiencias de antitrombina III, proteína C y S Un coágulo de sangre puede bloquear una arteria o una vena en el corazón, afectando: El corazón (angina de pecho o un ataque cardíaco) Los intestinos (isquemia mesentérica) o trombosis venosa mesentérica Los riñones (trombosis de la vena renal) Las arterias de las piernas o brazos Las piernas (trombosis venosa profunda) Los pulmones (embolia pulmonar) El cuello o el cerebro (accidente cerebrovascular) Los trombos y émbolos pueden fijarse firmemente a un vaso sanguíneo y pueden bloquear parcial o completamente el flujo de sangre a dicho vaso. Un bloqueo en el vaso sanguíneo impide que el flujo normal de sangre y oxígeno llegue a los tejidos en ese sitio, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede ocasionar daño tisular o necrosis en esa área. DIAPOSITIVA #7 FIBRINÓLISIS
Después de que el coágulo se ha establecido, comienza la reparación de los tejidos afectados con el proceso de cicatrización. Para hacer posible esto el coágulo es colonizado por células que formarán nuevos tejidos y en el proceso va siendo degradado. La degradación de la fibrina (fibrinólisis), componente mayoritaria del coágulo, es catalizada por la enzima plasmina, una serina proteasa que ataca las uniones peptídicas en la región triple hélice de los monómeros de fibrina. La plasmina se genera a partir del plasminógeno, un precursor inactivo; activándose tanto por la acción de factores intrínsecos (propios de la cascada de coagulación) como extrínsecos, el más importante de los cuales es producido por el endotelio vascular. Se le denomina "activador tisular del plasminógeno" (t-PA). El gen de este factor ha sido clonado y actualmente se puede obtener la proteína producida por tecnología de ADN recombinante. Este factor suele utilizarse en clínica para favorecer la disolución de trombos.
SISTEMA FIBRINOLITICO La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón. La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca: En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA. En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
PLAQUETA Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro, derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos. Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva.
Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebro vascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores; cualquier anormalidad o enfermedad de las plaquetas es denominada trombocitopatía, la cual puede ser, ya sea un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Hay desórdenes que pueden reducir el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos, pero en cambio otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragia.
Las plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular; Estos dos factores de crecimiento han demostrado jugar un papel significativo en la regeneración y reparación del tejido conectivo; Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.
DESCUBRIMIENTO Max Schultze (1825-1874), un anatomista alemán, marcó la historia del descubrimiento de las plaquetas. Sin embargo los glóbulos rojos, o eritrocitos, ya eran conocidos desde van Leeuwenhoek, Schultze fue primero en publicar una descripción de las plaquetas.11Él describió "esférulas" mucho más pequeñas que los eritrocitos que ocasionalmente se agrupaban y participaban en colecciones defibrina, recomendando estudios adicionales sobre estos hallazgos.
Giulio Bizzozero (1846-1901), aportó sobre los hallazgos de Schultze, usando "circulación en vivo" para estudiar las células sanguíneas de anfibios microscópicamente. Él notó especialmente que las plaquetas se agrupaban en el sitio de lesión vascular, un proceso que precedía a la formación de un coágulo. Esta observación confirmó el papel de las plaquetas en la coagulación.
CINÉTICA Las plaquetas son producidas en el proceso de formación de las células sanguíneas llamado (trombopoyesis) en la médula ósea, por fragmentación en los bordes citoplasmáticos de los megacariocitos. • El rango fisiológico de las plaquetas es de 150-400 x 109/litro. • Alrededor de 1 x 1011 plaquetas de media son producidas cada día por un adulto sano. • La expectativa de vida de las plaquetas circulantes es de 7 a 10 días. • La producción de megacariocitos y plaquetas está regulada por la trombopoyetina, una hormona producida usualmente por el hígado y los riñones. • Cada megacariocito produce entre 5.000 y 10.000 plaquetas. • Las plaquetas son destruidas por fagocitosis en el bazo y por las células de Kupffer en el hígado. • Una reserva de plaquetas es almacenada en el bazo y son liberadas cuando se necesitan por medio de contracción esplénica mediada por el sistema nervioso simpático.
Formación de trombos La función plaquetaria consiste en el mantenimiento del corazón; Esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio.
ACTIVACIÓN DE LAS PLAQUETAS La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP.
Las células endoteliales producen una proteína llamada factor de von Willebrand (FvW),un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas.
Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo. Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio. La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria(plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina yfosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
CAMBIO DE FORMA Las plaquetas activadas cambian su forma haciéndose más esféricas, y formando pseudopodos en su superficie. De esta forma toman una forma estrellada.
SECRECIÓN DE GRÁNULOS Las plaquetas contienen gránulos alfa y gránulos densos. Las plaquetas activadas excretan el contenido de estos gránulos dentro de sus sistemas canaliculares y en la sangre circundante. Existen dos tipos de gránulos: • Gránulos densos (contienen ADP o ATP, calcio, y serotonina) • Granulos-α (contienen factor 4 plaquetario, factor de crecimiento transformante beta 1 (TGF beta 1), factor de crecimiento derivado de plaquetas, fibronectina, B-tromboglobulina, FvW, fibrinógeno, y factores de coagulación factor V y XIII). SÍNTESIS DE TROMBOXANO A2 La activación plaquetaria inicia la vía del ácido araquidónico para producir Tromboxano A2; el tromboxano A2 está involucrado en la activación de otras plaquetas y su formación es inhibida por los inhibidores de la COX, como el ácido acetilsalicílico.
ADHESIÓN Y AGREGACIÓN La agregación plaquetaria, usa el fibrinógeno y el FvW como agentes conectores. El receptor de agregación plaquetaria más abundante es la glicoproteina IIb/IIIa (gpIIb/IIIa); se trata de un receptor para el fibrinógeno dependiente del calcio, fibronectina, vitronectina,trombospondina, y factor de von Willebrand (FvW). Otros receptores incluyen el complejo GPIb-V-IX (FvW) y GPVI (colágeno).ó Las plaquetas activadas se adherirán, via glicoproteína (GP) Ia, al colágeno expuesto por el daño epitelial. La agregación y adhesión actúan juntos para formar el tapón plaquetario. Los filamentos de Miosina y actina en las plaquetas son estimuladas para contraerse durante la agregación, reforzando todavía más el tapón.
La agregación plaquetaria es estimulada por el ADP, tromboxano, y la activación del receptor-α2, pero inhibido por agentes antiinflamatorios como las prostaglandinas PGI2 y PGD2. La agregación plaquetaria se ve aumentada por la administración exógena de esteroídes anabólicos.
REPARACIÓN DE HERIDAS El coágulo sanguíneo es solo una solución temporal para detener la hemorragia; la reparación del vaso debe ocurrir después. La agregación plaquetaria ayuda en este proceso mediante la secreción de sustancias químicas que promueven la invasión de fibroblastosdel tejido conectivo adyacente hacia el interior de la herida para formar una costra. El coágulo obturador es lentamente disuelto por la enzima fibrinolítica, plasmina, y las plaquetas son eliminadas por fagocitosis.
Otras funciones • Retracción del coágulo • Pro-coagulación • Inflamación • Señalización citoquínica • Fagocitosis
SEÑALIZACIÓN CITOQUÍNICA Adicionalmente a su función de ser el efector celular de la hemostasia, las plaquetas son rápidamente depositadas en sitios de lesión o infección, y potencialmente modulan los procesos inflamatorios por medio de la interacción con leucocitos y por la secreción decitoquinas, quimiosinas, y otros mediadores de la inflamación .las plaquetas también secretan factor de crecimiento derivado de plaquetas (PDGF).
PAPEL EN ENFERMEDADES Recuentos altos y bajos El recuento de plaquetas de un individuo sano se encuentra entre 150,000 y 450,000 por μl (microlitro) de sangre (150-450 x 109/L).18 El 95 % de los individuos sanos tendrán recuentos de plaquetas dentro de este rango. Algunos tendrán recuentos de plaquetas estadísticamente anormales sin tener ninguna anormalidad demostrable. Sin embargo, si el recuento es muy alto o muy bajo la probabilidad de que una anormalidad este presente es más alta. Tanto la trombocitopenia como la trombocitosis pueden manifestarse como problemas de coagulación. En general, los recuentos bajos de plaquetas incrementan el riesgo de sangrado; sin embargo existen excepciones. Por ejemplo la trombocitopenia inmune inducida por heparina.En la trombocitosis se puede producir trombosis, sin embargo esto sucede principalmente cuando el recuento elevado es debido a desordenes mieloproliferativos. Los recuentos de plaquetas en general, no son corregidos con transfusión a menos que el paciente esté sangrando o el recuento haya caído por debajo 5 x 109/L. La transfusión esta contraindicada en la púrpura trombocitopénica idiopática (PTI), puesto que estimula lacoagulopatía. En los pacientes sometidos a cirugía, niveles inferiores a 50 x 109/L están asociados a sangrado quirúrgico anormal, y procedimientos anestésicos regionales como la anestesia epidural son evitados para niveles inferiores a 80-100 x 109/L.
El recuento normal de plaquetas no es garantía de función adecuada. En algunos estados, las plaquetas, siendo normales en número, son disfuncionales. Por ejemplo, el ácido acetilsalicílico interrumpe irreversiblemente la función plaquetaria mediante la inhibición de laciclooxigenasa-1 (COX1), y por consiguiente la hemostasia normal. Las plaquetas resultantes no tienen ADN y son incapaces de producir nueva ciclooxigenasa. La función plaquetaria normal no se restaurara hasta que el uso de ASA haya cesado y un número suficiente de las plaquetas afectadas hayan sido reemplazadas por nuevas, lo cual suele tardar unos 7 días. El Ibuprofeno, un AINE, no tiene un período tan largo de efecto, y la función plaquetaria vuelve a la normalidad dentro de las 24 horas,19 y tomando ibuprofeno antes que el ASA prevendrá los efectos irreversibles de esta.20 La Uremia, a consecuencia de la insuficiencia renal, conduce a la disfunción plaquetaria que puede ser aminorada con la administración de desmopresina.
MEDICAMENTOS Agentes orales usados a menudo para alterar/suprimir la función plaquetaria: • Ácido acetilsalicílico • Clopidogrel • Cilostazol • Ticlopidina Agentes intravenosos usados a menudo para alterar/suprimir la función plaquetaria: • Abciximab • Eptifibatida • Tirofiban
ENFERMEDADES Desordenes que provocan un recuento bajo de plaquetas: • Trombocitopenia • Púrpura trombocitopénica idiopática • Púrpura trombocitopénica trombótica • Púrpura trombocitopenica inducida por medicamentos • Enfermedad de Gaucher • Anemia aplásica Trastornos Aloimunes • Trombocitopenia fetomaterna autoinmune • Algunas reacciones trasfusionales Desordenes que provocan disfunción o recuento reducido: • Síndrome HELLP • Síndrome urémico hemolítico • Quimioterapia • Dengue • Deficiencia del almacenamiento en gránulos Alfa-Delta; es un desorden hemorrágico hereditario.21 Desordenes caracterizados por recuentos elevados: • Trombocitosis, incluyendo trombocitosis esencial (recuento elevado, ya sea reactivo o como una expresión de trastorno mieloproliferativo); puede mostrar plaquetas disfuncionales. Desordenes de la agregación y adherencia plaquetarios: • Síndrome de Bernard-Soulier • Tromboastenia de Glanzmann • Síndrome de Scott's • Enfermedad de von Willebrand • Síndrome de Hermansky-Pudlak • Síndrome de plaquetas grises Desordenes del metabolismo de plaquetas • Actividad disminuida de la oxigenación, inducida o congénita • Defectos del almacenamiento, adquirido o congénito Desordenes que comprometen la función plaquetaria: • Hemofilia Desordenes en los cuales las plaquetas juegan un papel clave: • Aterosclerosis • Enfermedad coronaria arterial, e infarto del miocardio • Accidente cerebro vascular • Enfermedad arterial oclusiva periférica • Cancer
PLASMA RICO EN PLAQUETAS (PRP) La preparación del plasma rico en plaquetas (PRP) de procedencia autóloga, requiere la extracción y recolección de sangre periférica del paciente, la separación de las plaquetas y el plasma de los otros elementos formes sanguíneos, y la posterior polimerización de la fibrina de dicho plasma para concentrar las plaquetas formando un gel rico en plaquetas con suficiente estabilidad como para ser implantado quirúrgicamente. Actualmente, algunos métodos comerciales para la preparación del PRP utilizan calcio y trombina bovina o bien, trombina preparada de forma autóloga para crear una matriz rica en plaquetas y fibrina (PRFM del inglés- platelet-rich fibrin matrix). La preparación de trombina autóloga requiere tiempo y pasos adicionales, así como un mayor volumen de sangre; por otro lado, el uso de trombina bovina ha sido asociado con el desarrollo de anticuerpos contra los factores de coagulación V y XI, y la misma trombina, aumentando de esta forma el riesgo de anormalidades en la coagulación. Adicionalmente, para asegurar una degranulación de las plaquetas y la formación de un coágulo estable, se utilizan grandes cantidades de trombina, esto puede causar una liberación inmediata de los factores de crecimiento.
La liberación de factores de crecimiento es desencadenada por la activación de las plaquetas, esta puede ser iniciada por una gran variedad de sustancias o estímulos como la trombina, el cloruro de calcio, el colágeno o el adenosina 5c-difosfato.Un coágulo sanguíneo de PRP contiene aproximadamente un 4% de glóbulos rojos, 95% de plaquetas y 1% de glóbulos blancos. Las propiedades del PRP están basadas en los múltiples factores de crecimiento y de diferenciación producidos y liberados a raíz de la activación de las plaquetas. Estos factores son críticos para la regulación y estimulación del proceso curativo de lesiones. Por otro lado, existe una segunda generación del concentrado de plaquetas que recibe el nombre de matriz rica en plaquetas y fibrina (PRFM del inglés- platelet-rich fibrin matrix), la cual es un mejoramiento del PRP preparado tradicionalmente. Existe un método actual que evita el uso de trombina como activador. Este sistema utiliza únicamente calcio y centrifugación para activar la polimerización de la fibrina y formar así el PRFM. PRFM, en forma de gel o una membrana densa y flexible, puede ser aplicada al paciente y la liberación de los factores de crecimiento es desencadenada por los activadores autólogos presentes en el sitio de aplicación. Este método permite una liberación gradual de los factores de crecimiento en el sitio de aplicación, que pueden emitir señales a diferentes tipos celulares para que emitan una respuesta en momentos apropiados. Estudios in vitro indican que el PRFM presenta una liberación gradual y estable de los factores de crecimiento a lo largo de 7 días. El plasma rico en plaquetas (PRP) puede obtenerse por medio de diferentes técnicas ya sean separadores celulares de propósitos generales o bien, separadores celulares para la concentración de plaquetas. Muchos productos comerciales se encuentran disponibles en este campo, la mayoría de ellos obtienen resultados similares, cuyas diferencias se deben fundamentalmente al precio, tiempo, espacio requerido y la tecnología necesaria para fabricarlo. Son pocos los productos comerciales disponibles para la obtención de una matriz rica en plaquetas y fibrina como producto final.
COAGULACIÓN Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre. Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdaderocambio de estado.
Este proceso es debido, en última instancia, a que unaproteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregadosmacromoleculares en forma de una red tridimensional. El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimáticopor el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse. Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas. Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
FACTORES DE COAGULACIÓN El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc.
En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares. A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenossintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada. Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola. Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
ETAPAS DE LA CASCADA DE COAGULACIÓN La cascada de coagulación se divide para su estudio, clásicamente en tres vias: La via intrínseca, la vía extrínseca y la vía común.
Las vías intrínseca y extrínseca son las vías de iniciación de la cascada, mientras que la vía común es hacia donde confluyen las otras dos desembocando en la conversión de fibrinógeno en fibrina.
Esta división es un tanto arbitraria y tiene más que ver con las deficiencias de las técnicas que en su momento se utilizaron para desentrañar los mecanismos implicados, que con lo que ocurre realmente en una lesión vascular; ya que en este último caso se establecen varias interrelacciones entre las vías de iniciación.
Mecanismo básico Cada reacción de estas vías da como resultado el ensamblado de un complejo compuesto por una enzima (factor de coagulación activado), un sustrato (proenzima de un factor de coagulación) y un cofactor que actúa posibilitando la reacción.
Estos componentes se ensamblan en general sobre una superficiefosfolipídica y se mantienen unidos por medio de puentes formados por iones Ca2+. Por lo tanto la reacción en cascada tiende a producirse en un sitio donde este ensamblaje puede ocurrir; por ejemplo sobre la superficie de plaquetas activadas.
Tanto la vía intrínseca como la vía extrínseca desembocan en la conversión del factor X en Xa (la letra "a" como subíndice "a" significa "activado") punto en el que se inicia la vía común.
Vía intrínseca Recibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso hemostático.
El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular.
En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación.
En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como lacelulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción. A esta vía es posible subdividirla en tres pasos:
Formación del factor XIa En esta etapa participan cuatro proteínas: Precalicreína, Quininógeno de alto peso molecular (HMWK) y los factores XII y XI. Esta etapa no requiere de iones calcio.
Estos cuatro factores se adsorben sobre la superficie cargada negativamente, formando el complejo cebador o de iniciación. De estos factores el XII funciona como verdadero iniciador, ya que si bien es una proenzima, posee una pequeña actividad catalítica que alcanza para activar a la precalicreína convirtiéndola en calicreína. En segunda instancia la calicreína actúa catalíticamente sobre el factor XII para convertirlo en XIIa, una enzima muchísimo más activa. La actividad catalítica de la calicreína se ve potenciada por el HMWK. Por último la proteasa XIIa actúa sobre el factor XI para liberar XIa.
Formación del factor IXa El factor IX se encuentra en el plasma como una proenzima. En presencia de iones Ca2+ el factor XIa cataliza la ruptura de una unión peptídica en la molécula del factor IX para formar un glucopéptido de 10 KDa y liberar por otro lado al factor IXa. El factor IX se encuentra ausente en personas con hemofilia tipo B.
Formación del factor Xa Sobre la membrana de las plaquetas se forma un complejo constituido por los factores IXa, X y VIII.
Los residuos gamma-carboxiglutamato de los factores IXa y X actúan como quelantes del ion Ca2+, permitiendo que estos componentes formen un complejo unido por medio de puentes de iones calcio y ayudando a que el complejo se ancle a los fosfolípidos de membrana. Primero se unen los factores X y IXa a la membrana y luego se une el VIII.
El factor VIII es en realidad un homorodímero, formado por cuatro cadenas proteicas, cada una codificada por un gen diferente (VIII:C y VIII:R). El componente VIII:C es conocido como "componente antihemofílico" y actúa como cofactor del IXa en la activación del factor X, el componente VIII:R es el que permite la unión del factor VIII al complejo. La ausencia del componente antihemofílico causa hemofilia A.
El complejo formado por los factores IXa-X-VIII-Fosfolípidos y Ca2+ actúa sobre el factor X para convertirlo en Xa. En este punto concluye la vía intrínseca.
Vía extrínseca Recibió este nombre debido a que fue posible notar desde un primer momento que la iniciación de esta vía requería de factores ajenos a la sangre.
Cuando la sangre entra en contacto con tejidos lesionados o se mezcla con extractos de tejidos, se genera muy rápidamente factor Xa. En este caso la activación de la proenzima X es mediada por un complejo formado por factor VII, Ca2+ y factor tisular unido afosfolípidos provenientes de las membranas celulares rotas y de las plaquetas (antiguamente este complejo factor tisular-fosfolípidos era conocido como tromboplastina).
El factor tisular es una lipoproteína sintetizada en el endotelio de los vasos sanguíneos de todos los tejidos, aunque es especialmente abundante en pulmón, cerebro y placenta. El factor tisular se encuentra normalmente "secuestrado" en el interior de las células endoteliales y es secretado en respuesta a una lesión, o bajo el efecto de algunas citoquinas tales como el Factor de Necrosis Tumoral (TNF), InterLeucina 1 (IL-1); o por endotoxinas bacterianas.
La vía extrínseca es muy rápida, se cumple en apenas unos segundos y comprende dos pasos; mientras que la intrínseca insume varios minutos.
Formación del factor VIIa En primera instancia el factor VII se une a la porción fosfolipídica del factor tisular gracias a sus residuos gamma-carboxiglutamato, utilizando iones Ca2+ como puentes. Este complejo provoca la activación del factor VIIa.
Formación del factor Xa El complejo VIIa-III-Ca2+ actúa sobre el factor X convirtiéndolo en la proteasa activa Xa. En este punto termina la vía extrínseca y se inicia la vía común
Vía común Llegando al punto en que se activa el factor X, ambas vías confluyen en la llamada vía común. La vía común termina con la conversión de fibrinógeno en fibrina, y el posterior entrecruzamiento de la misma estabilizando el coágulo.
La vía común implica tres etapas: Formación de trombina La trombina (también llamada factor II a) es una proteasa generada por la ruptura de la cadena proteica de la proenzima protrombina (factor II), una glicoproteína constituida por 582 aminoácidos y con 12 puentes disulfuro intracatenarios.
La trombina se activa luego de que la proteasa Xa hidroliza dos uniones peptídicas de la protrombina. La Xa produce en primer término la escisión de un fragmento de 32 KDa de la región N-terminal de la cadena, cortándola sobre una unión arginina-treonina. En segundo término produce la ruptura de un enlace entre una arginina y una isoleucina; sin embargo estos dos últimos fragmentos permanecen unidos por un puente disulfuro.
La trombina es una serina-proteasa similar a la tripsina, pero mucho más selectiva. Ataca casi de manera exclusiva las uniones arginina con un aminoácido cargado positivamente en sus sustratos.
La conversión de protrombina a trombina debida al factor Xa se acelera notablemente por la formación de un complejo con el factor Va y Ca2+ sobre la superficie de las membranas plaquetarias (fosfolípidos de membrana).
El factor Xa y la protrombina se adsorben sobre la membrana utilizando iones Ca2+como puentes. El factor Va se une a la protrombina acelerando la reacción.
El factor Va se produce por la acción de la trombina sobre el factor V en un claro ejemplo de una reacción que va acelerándose a medida que progresa (reacción autoacelerada).
Formación de fibrina El fibrinógeno (factor I) es una glicoproteína compuesta por seis cadenas polipeptídicas: dos A-alfa, dos B-beta y dos gamma; unidas entre sí por puentes disulfuro.
Se trata de una molécula alargada y simétrica formada por tres dominios globulares conectados por segmentos fibrilares. Cada mitad de la molécula se encuentra formada por tres cadenas (A-alfa, B-beta y gamma) que se enrollan en una triple hélice muy compacta en los sectores fibrilares. Los extremos amino de las seis cadenas se reúnen en el dominio globular central.
En un hecho que parecería muy curioso, los extremos N-terminales de las cadenas A-alfa y B-beta emergen como cabos libres del dominio globular central.
Estas cadenas son muy ricas en aspartato y glutamato, además las cadenas B-beta poseeen en esta región residuos tirosina-O-sulfato formados postraduccionalmente. Estos residuos con una alta tendencia a adquirir carga negativa contribuyen a formar una región central con una muy alta densidad de carga. Esta región electronegativa central es la responsable de la repulsión entre moléculas de fibrina que las mantiene en solución.
La trombina ataca los enlaces arginina-glicina presentes en estos "cabos libres", separando cuatro péptidos; dos segmentos A de 18 aminoácidos cada uno (provenientes de las cadenas A-alfa), y dos segmentos B de 20 aminoácidos (provenientes de las cadenas B-beta). A estos péptidos se los suele denominar "fibrinopéptidos". El resto que queda de la molécula es un monómero de fibrina de composición alfa2beta2gamma2.
Al eliminarse los fibinopéptidos desaparecen las fuerzas de repulsión intermoleculares con lo que los monómeros de fibrina tienden a agruparse espontáneamente formando asociaciones altamente ordenadas.
Los monómeros se disponen uno a continuación del otro, cabeza con cabeza en forma de largas hebras. Estas hebras a su vez forman manojos, emparejándose con otras hebras de tal manera que la región central de los monómeros de fibrina de una se encuentra rodeada por las cabezas de los monómeros de fibrina de las otras.
Este emparejamiento se hace posible gracias a interaciones de tipo electrostático y puente hidrógeno entre las regiones centrales de los monómeros de una y las cabezas globulares de otras.
Entrecruzamiento de la fibrina Los haces paralelos de fibrina polimerizada forman una asociación laxa, que se encuentra en equilibrio con la forma monomérica de la molécula; por lo que sería imposible que cumplieran su papel de formar un coágulo estable sin reforzar esta estructura por medio de enlaces covalentes entre hebras vecinas.
La formación de estos "puentes" covalentes intercatenarios es catalizada por la enzima transglutaminasa (conocida también como factor XIIIa).
La transglutaminidasa cataliza la formación de enlaces amida entre restos glutamina y lisina de hebras próximas entre sí. En la reacción se libera amoníaco en forma de ion amonio (NH4+). Esta enzima se forma a partir del factor XIII por acción de la trombina.
Regulación y modulación de la cascada Debido a que la cascada de coagulación consiste en una serie de reacciones que van amplificándose y acelerándose en cada paso, es lógico pensar que debe existir algún mecanismo de regulación; un "freno" a la reacción en cadena; ya que de progresar sin control en pocos minutos podría provocar un taponamiento masivo de los vasos sanguíneos (trombosis diseminada).
Varios mecanismos intervienen en la regulación de la cascada de reacciones: • El flujo sanguíneo normal, arrastra a los factores activados, diluyendo su acción e impidiéndoles acelerarse. Esta es una de las razones por las cuales cuando existe estasis del flujo sanguíneo se favorece la formación de trombos. • El hígado actúa como un filtro quitando de la sangre en circulación los factores activados e inactivándolos. • Existen además algunas proteasas que degradan específicamente a ciertos factores activados, y otras que ejercen acciones inhibitorias sobre factores activos.
Proteína C La proteína C es una proenzima que se encuentra normalmente en el plasma, y cuya síntesis en el hígado es dependiente de la vitamina K. Esta proteína es convertida en una proteasa activa por la acción de la trombina. La proteína Ca actúa específicamente degradando a los factores Va y VIIIa, con lo que limita la proyección de la cascada.
Es interesante notar el triple papel que desempeña la trombina: cataliza la formación de fibrina, activa a la enzima responsable de su entrecruzamiento, y una vez que el proceso de coagulación y estabilización del coágulo está en marcha; ejerce acciones tendientes a limitarlo.
Antitrombina III La antitrombina III es una glicoproteína de 60 Kda sintetizada en el hígado sin depender de la vitamina K, es considerada la principal inhibidora de la coagulación. Esta proteína actúa inhibiendo irreversiblemente a varios factores procoagulantes activos, el principal de los cuales es la trombina; aunque también actúa sobre la calicreína y los factores IXa, Xa, XIa y XIIa.
La acción de la antitrombina es notablemente aumentada por el heteropolisacárido heparina. La heparina se encuentra en el endotelio de los vasos sanguíneos y en los gránulos de las células cebadas, tiene una poderosa acción anticoagulante ya que facilita la unión de la antitrombina III con los factores procoagulantes activos.
Existen otras anti-proteasas sanguíneas que también ejercen acción anticoagulante aunque menos potente tales como la alfa2macroglobulina y la alfa1 antitripsina.
Anticoagulantes Un anticoagulante es, como su nombre lo indica, una sustancia química que retrasa o impide la coagulación de la sangre, ya sea en el interior de un organismo (In Vivo) o en el exterior (In Vitro) Existen diferentes tipos de anticoagulantes que actúan dificultando o impidiendo alguno de los pasos de la cascada de coagulación.
Existen dos tipos principales de anticoagulantes, los anticoagulantes para uso "In Vitro" y los que tienen empleo "In Vivo", entre estos últimos se encuentran los medicamentos con acción anticoagulante.
En general los anticoagulantes para uso In Vitro actúan como quelantes del ion Ca2+, de manera tal que este no puede participar en la formación de los complejos que activan al factor X, y por lo tanto se interrumpe la cascada de coagulación casi en su inicio. Los anticoagulantes para uso In Vivo actúan de maneras un poco más complicadas. La adición de algunos agentes quelantes tales como el EDTA entrañan un grave riesgo para la salud del individuo sometido a tratamiento, ya que estos agentes son capaces de acomplejar gran cantidad de iones con alta afinidad, algunos de los cuales desempeñan importantes funciones en el organismo tales como el Cu2+, Fe3+, Zn2+, etc; mientras que otros agentes acomplejantes del calcio tales como el citrato, no tienen gran utilidad ya que son rápidamente metabolizados perdiendo su capacidad anticoagulante. Entre los anticoagulantes para uso in vivo encontramos sustancias tales como la heparina o los anticoagulantes dicumarínicos.
Para uso In Vitro EDTA (C10H16N2O8) o sal disódica, dipotásica o tripotásica del ácido etilendiaminotetraacético. Esta sustancia actúa mediante un efecto quelante sobre el ion calcio (Ca2+, lo que impide la formación de los complejos procoagulantes en los que este ion participa. Este anticoagulante se utiliza fundamentalmente para la realización de recuentos celulares, sobre todo en autoanalizador. Tiene la ventaja de permitir la realización del hematocrito y de frotis sanguíneo hasta dos horas después de la extracción de la muestra. También impide la aglutinación de las plaquetas. • Heparina Sódica. Heparina de Litio, es un anticoagulante fisiológico que actúa impidiendo que la protrombina se transforme en trombina. Estructuralmente es un mucopolisacárido ácido que posee grupos sulfato. Esta última característica no la hace adecuada para muestras que van a ser examinadas al microscopio luego de tinción, ya que altera notablemente las coloraciones obtenidas. • Citrato Trisódico (C6H5O7Na3) actúa impidiendo que el calcio se ionice, evitando así la coagulación. Se utiliza principalmente para realizar pruebas de hemostasia; así como también para medir la velocidad de eritrosedimentación. • ACD, es un anticoagulante formado por una mezcla de compuestos (Ácido citrico Citrato y Dextrosa en una proporción de 0.9, 2 y 2 g respectivamente en 120 ml de agua destilada) se emplea fundamentalmente en bancos de sangre para conservar las unidades de sangre y para realizar estudios metabólicos eritrocitarios ya que permite una buena conservación de los hematíes.
Anticoagulantes para uso In Vivo (medicamentos anticoagulantes) Este grupo de anticoagulantes se definen como "medicamentos que impiden la coagulación o la agregación plaquetaria" Este tipo de medicamentos tienen utilidad en aquellas patologas causadas por un trombo sanguíneo, ya sea para facilitar su disolución (trombolisis) o bien para prevenir que los trombos se repitan.
En este artículo vamos a centrarnos en aquellos que impiden la cascada de coagulación: • La heparina alarga el tiempo de coagulación, se administra generalmente mediante inyección subcutánea o endovenosa. Ya que es un compuesto fisiológico presente en gran cantidad en los mamíferos, comúnmente se utiliza heparina obtenida de pulmón de vaca o de mucosa intestinal de cerdo convenientemente purificada. La potencia difiere según el origen, pero hoy en día vienen estandarizadas en UI, por lo que se pueden comparar solo con este índice.
Comercialmente se obtiene en forma de dos sales (cálcica y sódica) que no guardan demasiada diferencia en su actividad. Las cálcicas se usan preferentemente por vía subcutánea, ya que resultan menos dolorosas, pero por vía endovenosa pueden utilizarse ambas. La heparina NUNCA se administra vía intramuscular.
La Heparina se utiliza cuando se precisa de acción anticoagulante rápida y por poco tiempo. En la prevención de trombosis venosas de cirugía se utiliza a bajas dosis, 5.000UI, dos horas antes de la intervención y después cada 12 horas hasta el alta del paciente. Las heparinas de bajo peso molecular son fragmentos de peso molecular entre 3.500 y 6.000, con ello tiene una vida más larga y aumenta su biodisponibilidad. Tiene una menor inhibición de la agregación plaquetaria. No sustituyen a las heparinas tradicionales sino que en terapias de baja dosis son más cómodas porque se aplican una sola vez al día.
En terapias de altas dosis se utilizan las heparinas tradicionales. • Hirudina • Anticoagulantes dicumarínicos. Reciben este nombre genérico un grupo de compuestos derivados del Dicumarol (un compuesto extraído del trébol dulce) entre los que se encuentran el Acenocumarol (el de uso más frecuente en España, bajo el popular nombre de Sintrom) y la Warfarina. Estos medicamentos presentan la ventaja de poder ser administrados por vía oral y de poseer un efecto prolongado en el tiempo, con gran variabilidad interindividual, por ello necesitan controles periódicos para su ajuste terapéutico.
Todos ellos son inhibidores de la vitamina K (aVK). Debido a que la vitamina K interviene como cofactor enzimático en la síntesis de los factores II,VII,IX y X (concretamente en la gamma-carboxilación de estos); el resultado es que provoca la aparición en sangre, de unas formas inactivas de los mismos denominadas PIVKAs (“Proteins Induced by Vitamin K Antagonists”).
Dada la diferente vida media que presentan los factores de coagulación (el tiempo que permanecen en sangre antes de ser degradados), por ejemplo el VII comienza a descender en 6 horas pero el II tarda cerca de 70, no se consigue una anticoagulación efectiva hasta el 3º-4º día de tratamiento y el efecto no se estabiliza hasta después de una semana.
Curioso es que la activación de dos inhibidores fisiológicos de la coagulación como son las Proteínas C y S de importancia fundamental (inhiben a los Factores V y VIII activados), también depende de la Vit. K, por lo que los cumarínicos originan una “paradoja bioquímica” anticoagulante-procoagulante.
No obstante, su efecto anticoagulante supera ampliamente al procoagulante, por lo que solo puede tener consecuencias clínicamente significativas en raros casos (Déficits congénitos de Proteína C o S) y de forma transitoria al inicio del tratamiento.
TROMBOS Y COÁGULOS Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida. • Un coágulo sanguíneo que se forma dentro de una de las venas o las arterias se denomina trombo y también se puede formar en el corazón. • Un trombo que se desprende y viaja desde un lugar en el cuerpo a otro se llama émbolo. Un trombo o émbolo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo. • Una obstrucción en una arteria puede impedir que el oxígeno llegue a los tejidos en esa área, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede provocar daños en los tejidos o la muerte. • Una obstrucción en una vena generalmente provocará acumulación de líquido e hinchazón.
Causas Las situaciones en las que es más probable que se forme un coágulo de sangre abarcan: • Estar en reposo en cama por largo tiempo. • Cruzar las piernas durante largos periodos al estar sentado o sentarse durante mucho tiempo, como en un avión o en un vehículo. • Durante y después del embarazo. • No tener suficiente agua en el cuerpo (deshidratación). • Tomar píldoras anticonceptivas u hormonas estrógeno (especialmente en las mujeres que fuman). • Utilizar un catéter intravenoso por largo tiempo.
Los coágulos de sangre también son más probables en personas con cáncer, cirugía o lesión recientes, obesidad y enfermedades del hígado o del riñón.
Una acumulación de colesterol que estreche una arteria puede cambiar o reducir el flujo de sangre, facilitando la formación de un coágulo sanguíneo o de un trombo.
Las afecciones que se transmiten de padres a hijos (hereditarias) pueden hacerlo más propenso a la formación de coágulos sanguíneos anormales. Los trastornos hereditarios que afectan la coagulación son: • Trombofilia por factor V de Leiden • Mutación de la protrombina G20210A • Otras afecciones raras como deficiencias de antitrombina III, proteína C y S Un coágulo de sangre puede bloquear una arteria o una vena en el corazón, afectando: • El corazón (angina de pecho o un ataque cardíaco) • Los intestinos (isquemia mesentérica) o trombosis venosa mesentérica • Los riñones (trombosis de la vena renal) • Las arterias de las piernas o brazos • Las piernas (trombosis venosa profunda) • Los pulmones (embolia pulmonar) • El cuello o el cerebro (accidente cerebrovascular)
Posibles complicaciones Los trombos y émbolos pueden fijarse firmemente a un vaso sanguíneo y pueden bloquear parcial o completamente el flujo de sangre a dicho vaso. Un bloqueo en el vaso sanguíneo impide que el flujo normal de sangre y oxígeno llegue a los tejidos en ese sitio, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede ocasionar daño tisular o necrosis en esa área.
FIBRINÓLISIS La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos.
Bioquímica La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasasen los macrófagos del hígado y el riñón.
La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca: • En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o lasinfecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógenoo tPA. • En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno.
Regulación La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Después de que el coágulo se ha establecido, comienza la reparación de los tejidos afectados con el proceso de cicatrización. Para hacer posible esto el coágulo es colonizado por células que formarán nuevos tejidos y en el proceso va siendo degradado.
La degradación de la fibrina (fibrinólisis), componente mayoritaria del coágulo, es catalizada por la enzima plasmina, una serina proteasa que ataca las uniones peptídicas en la región triple hélice de los monómeros de fibrina.
La plasmina se genera a partir del plasminógeno, un precursor inactivo; activándose tanto por la acción de factores intrínsecos (propios de la cascada de coagulación) como extrínsecos, el más importante de los cuales es producido por el endotelio vascular. Se le denomina "activador tisular del plasminógeno" (t-PA).
El gen de este factor ha sido clonado y actualmente se puede obtener la proteína producida por tecnología de ADN recombinante. Este factor suele utilizarse en clínica para favorecer la disolución de trombos.
SISTEMA FIBRINOLITICO El sistema fibrinolítico va a jugar además un papel importante en diversas situaciones en las que se produce proteolisis tisular, tales como inflamación, invasión tumoral o neovascularización. La importancia fisiopatológica del sistema fibrinolítico deriva de que las alteraciones que condicionan un defecto de actividad fibrinolítica pueden predisponer a la trombosis, mientras que un exceso de activación favorecería la aparición de hemorragia. Además va a tener una gran importancia en el área terapéutica por la aplicación creciente de fármacos fibrinolíticos en el tratamiento de la trombosis, especialmente en el infarto agudo de miocardio (IAM). Principales componentes del sistema fibrinolítico: La fibrinolisis es un encadenamiento (proceso) enzimático compuesto por una serie de activadores e inhibidores los cuales regulan la conversión del proenzima circulante, plasminógeno, en el enzima activo plasmina. La producción de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina y esto es muy importante para el mantenimiento de la permeabilidad vascular.
activacion plaquetaria Los procesos de activación plaquetaria comprenden complejas rutas de señalización que involucran sistemas de proteínas G acoplados a enzimas efectoras como la fosfolipasa A2, fosfolipasa C, adenilato y guanilato ciclasas. Las propiedades funcionales y estructurales de las plaquetas están estrechamente relacionadas, lo que les confiere un alto potencial trombogénico, contribuyendo sustancialmente en el desarrollo de un infarto cerebral. Se realizó una revisión de las bases de la agregación, activación, degranulación plaquetaria y su participación en la enfermedad cerebrovascular. Se realizó una actualización del uso de los agentes antiplaquetarios y de la literatura actual referente al uso de bloqueadores de las glicoproteínas adhesivas de membrana. Palabras clave: Activación plaquetaria, Infarto cerebral, Glicoproteína IIb/IIIa, Antiagregantes plaquetarios.
dilenny quezada 83876 cuagulacion de la sangre 1 Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre. Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional. El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse. Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas. Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo
dilenny quezada 83876 trombo y cuagulo Trombo: es una masa intravascular o intracardíaca formada por elementos de la sangre e insolubles en ella, se caracterizan por estar fijos a la pared del vaso por una zona donde son más rígidos y consistentes. Pueden ser arteriales o venosos.
Coagulos: intravasculares son postmorten son gelatinosos, con una porción declive rojo oscura porque los eritrocitos sedimentan por acción de la graverdad y un sobrenadante amarillo en grasa de pollo, no estan fijos a la pared del vaso. En cambio los trombos son mas consistentes, estan anclados en un punto a la pared del vaso o cardiaca, los arteriales consistentes de color blanco grisaceo y friables, formados por una red de plaquetas fibrina y leucocitos en degeneración (se generan en zonas donde hay turbulencia, como en la bifircación de los vasos) lo venosos son rojos y se desarrollan en zonas de estasis sanguineo por lo que retienen mayor número de hematíes, por eso son rojos...
dilenny quezada 83876 fibrinolisis La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón. La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca: En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA. En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno.
dilenny quezada 83876 sistema fibrinolitico REGULACIÓN DEL SISTEMA FIBRINOLÍTICO La hemostasia fisiológica asegura la permeabilidad vascular y la circulación sanguínea gracias a un complejo balance entre los mecanismos de coagulación, encargados de la formación de fibrina, y los de fibrinolisis responsables de su eliminación del torrente circulatorio. La fibrinolisis es un proceso específico de disolución de fibrina por proteasas sanguíneas, siendo la plasmina el enzima responsable de dicha degradación. La activación del sistema fibrinolítico es esencial para eliminar depósitos intravasculares de fibrina resultantes de la activación fisiológica o patológica del sistema de coagulación. El sistema fibrinolítico va a jugar además un papel importante en diversas situaciones en las que se produce proteolisis tisular, tales como inflamación, invasión tumoral o neovascularización (3, 4). La importancia fisiopatológica del sistema fibrinolítico deriva de que las alteraciones que condicionan un defecto de actividad fibrinolítica pueden predisponer a la trombosis, mientras que un exceso de activación favorecería la aparición de hemorragia. Además va a tener una gran importancia en el área terapéutica por la aplicación creciente de fármacos fibrinolíticos en el tratamiento de la trombosis, especialmente en el infarto agudo de miocardio (IAM).
Principales componentes del sistema fibrinolítico: La fibrinolisis es un encadenamiento (proceso) enzimático compuesto por una serie de activadores e inhibidores los cuales regulan la conversión del proenzima circulante, plasminógeno, en el enzima activo plasmina. La producción de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina (5) y esto es muy importante para el mantenimiento de la permeabilidad vascular (Fig. 1). Los principales componentes del sistema fibrinolítico se resumen en la Tabla I.
activacion plaquetaria El factor de activación plaquetaria es un fosfolípido de síntesis endógena relacionado directamente con diversos procesos como isquemia, necrosis, trombosis y otros. El principal blanco de sus efectos deletéreos se encuentra en las células endoteliales, leucocitos y plaquetas, donde provoca graves perturbaciones mecánicas, reológicas y bioquímicas en la unidad circulatoria. Se realizó una revisión bibliográfica del factor de activación plaquetaria, su síntesis endógena, sus funciones biológicas, con especial énfasis en su relación con el daño oxidativo, así como los principales antagonistas conocidos. Se enfatizó el efecto de los ginkgólidos contenidos en el EGB 761 (extracto de Ginkgo biloba) y su aplicación en la clínica de diversos procesos patológicos relacionados con la circulación cerebral y periférica.
mirian sujeidy marrero 85500 cuagulacion de la sangre 1 Son un grupo de afecciones en las cuales hay un problema con el proceso de coagulación sanguínea del cuerpo. Estos trastornos pueden llevar a que se presente sangrado intenso y prolongado después de una lesión. El sangrado también puede iniciarse de manera espontánea Causas
La coagulación normal de la sangre involucra hasta 20 proteínas plasmáticas diferentes, conocidas como factores de la coagulación o factores de la coagulación sanguínea. Estos factores interactúan con otros químicos para formar una sustancia llamada fibrina que detiene el sangrado.
Se pueden presentar problemas cuando faltan ciertos factores de la coagulación o éstos están muy bajos. Los problemas de sangrado pueden ir desde leves hasta severos.
Algunos trastornos hemorrágicos están presentes desde el nacimiento y se transmiten de padres a hijos (hereditarios). Otros se desarrollan por:
•Enfermedades como deficiencia de vitamina K y enfermedad hepática severa •Tratamientos como el uso de medicamentos para detener los coágulos de sangre (anticoagulantes) o el uso prolongado de antibióticos
mirian sujeidy marrero 85500 cuagulacion de la sangre 2 Es un problema de coagulación de la sangre que ocurre cuando hay una falta de una sustancia (protrombina) que se necesita para que la sangre coagule Cuando uno sangra, el cuerpo inicia una serie de reacciones que ayudan a que la sangre se coagule, lo cual se denomina cascada de la coagulación. El proceso involucra proteínas especiales, llamadas factores de coagulación. Cuando falta uno o más de estos factores de coagulación, generalmente hay una probabilidad más alta de sangrado.
Este trastorno ocurre cuando el cuerpo no tiene suficiente factor II, una importante proteína en la coagulación de la sangre. La deficiencia del factor II que se transmite de padres a hijos (hereditaria) es muy poco frecuente. Ambos padres deben ser portadores para transmitírselo a sus hijos. El hecho de tener un antecedente familiar de un trastorno hemorrágico es un factor de riesgo potencial. Con mucha frecuencia, la deficiencia del factor II es causada por:
Falta (deficiencia) de vitamina K por el uso prolongado de antibióticos, obstrucción de las vías biliares y absorción inadecuada de los nutrientes por parte del tracto intestinal. (malabsorción intestinal). Enfermedad hepática grave. Uso de medicamentos para prevenir la coagulación (anticoagulantes como warfarina o Coumadin). examenes Análisis del factor II Tiempo parcial de tromboplastina Tiempo de protrombina (TP)
mirian sujeidy marrero 85500 cuagulacion de la sangre 3 La Hemostasis es el proceso mediante el cual un organismo evita las pérdidas excesivas de sangre cuando hay daño en algún vaso sanguíneo. Una herida en el cuerpo causa vasoconstricción acompañada de la adhesión de plaquetas y la formación de fibrinas. La activación de las plaquetas conlleva la formación de un tapón haemostatico, que para el sangrado y después es reforzado por la fibirina. La importancia y velocidad de cada proceso depende del tipo de vena, arteria o capilar que haya sido lastimado. Los tapones haemostáicos en situaciones patológicas son conocidos como trombos y siempre ocurren in vivo, mientras que los coágulos son formaciones en sangre estática o in vitro.1
Cuadro La Trombosis es la formación patológica de tapones hemostaticos en el sistema vascular sin la presencia de sangrado. Existen tres principales factores de disposición a la trombosis, que incluyen el daño a la pared vascular de un vaso sanguíneo o su erosión, un flujo sanguíneo alterado (ya sea por factores genéticos como mal funcionamientos o ambientales como el estilo de vida) o una coagulabilidad anormal de la sangre como sucede en las últimas fases del embarazo o mientras se lleva un tratamiento con anticonceptivos orales.2 La coagulación de la sangre es el proceso encargado de convertir la sangre en una sustancia gelatinosa. El evento principal es la conversión de fibrinógenos solubles a tiras insolubles de fibrinas mediante la acción de la trombina, y también es el último paso de esta cascada enzimática. Los componentes o factores de coagulación están siempre presentes en la sangre como precursores inactivos de enzimas proteolíticas y cofactores. Se activan mediante proteólisis, y su forma activa se denomina con el sufijo a. En esta cascada, la activación de una pequeña cantidad de factores cataliza la activación de grandes cantidades de otros y así sucesivamente, consecuentemente esta cascada provee un mecanismo de amplificación. La cascada está controlada con inhibidores para evitar la solidificación de toda la sangre del cuerpo. Uno de los inhibidores más importantes es la antitrombina III que neutraliza las proteasas serínicas de la cascada (XIa, Xa, IXa y IIa). El endotelio vascular también ayuda a limitar la zona de coagulación.1 Los factores de coagulación y sus funciones principales se muestran a continuació
mirian sujeidy marrero 85500 trombo y coagulo El trombo es un coágulo sanguíneo que se forma en un vaso y permanece allí. La embolia es un coágulo que se desplaza desde el sitio donde se formó a otro lugar en el cuerpo. El trombo o embolia puede producirse en un vaso sanguíneo y obstruir el flujo sanguíneo en ese lugar, impidiendo el suministro de oxígeno y flujo sanguíneo a los tejidos circundantes. Esto puede ocasionar un daño, destrucción (infarto) e incluso la muerte o necrosis de los tejidos que se encuentran en esa área.
Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida.
Un coágulo sanguíneo que se forma dentro de una de las venas o las arterias se denomina trombo y también se puede formar en el corazón. Un trombo que se desprende y viaja desde un lugar en el cuerpo a otro se llama émbolo. Un trombo o émbolo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo.
Una obstrucción en una arteria puede impedir que el oxígeno llegue a los tejidos en esa área, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede provocar daños en los tejidos o la muerte. Una obstrucción en una vena generalmente provocará acumulación de líquido e hinchazón. Las situaciones en las que es más probable que se forme un coágulo de sangre abarcan:
Estar en reposo en cama por largo tiempo. Cruzar las piernas durante largos periodos al estar sentado o sentarse durante mucho tiempo, como en un avión o en un vehículo. Durante y después del embarazo. No tener suficiente agua en el cuerpo (deshidratación). Tomar píldoras anticonceptivas u hormonas estrógeno (especialmente en las mujeres que fuman). Utilizar un catéter intravenoso por largo tiempo. Los coágulos de sangre también son más probables en personas con cáncer, cirugía o lesión recientes, obesidad y enfermedades del hígado o del riñón.
mirian sujeidy marrero 85500 fibrinolisis . Disolución de la fibrina y, por extensión, disolución de un coágulo sanguíneo (trombólisis). Es un fenómeno que sobreviene normalmente algunos días o algunas semanas después de la formación del coágulo. Cuando se produce demasiado rápidamente, puede provocar hemorragias espectaculares (fibrinólisis hemorrágica) intra o postoperatorias (particularmente en Cirugía Torácica), o después de un parto, de un aborto o de un choque traumático; o bien puede provocar hemorragias menos importantes (equimosis, hematuria) en el curso de ciertos cánceres (próstata, páncreas, estómago), de las cirrosis y de las leucosis. Obedece a la liberación de activadores de la profibrinolisina, o bien a la producción de un fermento proteolítico por los tejidos enfermos. Puede ser primitiva aunque por lo general es secundaria, reactiva, en el curso de un síndrome de coagulación intravascular diseminada La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
LADY BAEZ 75261 Activacion plaquetaria La sangre circula por el interior de los vasos, los glóbulos rojos (la más abundante masa celular sanguínea) lo hace por el centro del vaso, mientras que las plaquetas circulan más próximas a la superficie endotelial para vigilar la integridad del endotelio. El endotelio normal proporciona una superficie no adhesiva para las plaquetas. Cuando se rompe el endotelio, el colágeno expuesto favorece la adhesión de las plaquetas por medio de la interacción con una cantidad de receptores, como vemos en el video, pero es esencial para concretar la adhesión el Factor de Von Willebrand. A partir de esto y con el aumento del Ca++ intracelular se produce la activación de las plaquetas, lo que genera una serie de eventos que favorece la agregación plaquetaria, como son: -Cambio de forma y de ser una esfera regular pasa a tener prolongaciones. -Liberación del contenido de los gránulos (degranulación): ADP que torna a las plaquetas pegajosas,TX A2 derivado del Ácido Araquidónico (vasoconstrictor y agreganteplaquetario) actor II (trombina). Adhesión, Activación y Agregación son las fases que participan en la formación del tapón plaquetario . Proceso de coagulacion Los factores de coagulación son proteínas de la sangre que controlan el sangrado. Cuando un vaso sanguíneo se lesiona, sus paredes se contraen para limitar el flujo de sangre al área dañada. Entonces, pequeñas células llamadas plaquetas se adhieren al sitio de la lesión y se distribuyen a lo largo de la superficie del vaso sanguíneo. Al mismo tiempo, pequeños sacos al interior de las plaquetas liberan señales químicas para atraer a otras células al área y hacer que se aglutinen a fin de formar lo que se conoce como tapón plaquetario. En la superficie de estas plaquetas activadas muchos factores de coagulación diferentes trabajan juntos en una serie de reacciones químicas complejas (conocidas como cascada de la coagulación) para formar un coágulo de fibrina. El coágulo funciona como una red para detener el sangrado. Los factores de la coagulación circulan en la sangre sin estar activados. Cuando un vaso sanguíneo sufre una lesión se inicia la cascada de la coagulación y cada factor de la coagulación se activa en un orden específico para dar lugar a la formación del coágulo sanguíneo. Los factores de la coagulación se identifican con números romanos (e. g. factor I o FI). Fibrinolisis La fibrinólisis primaria se refiere a la descomposición normal de los coágulos, mientras que la fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa. Consecuencias Cuando se produce demasiado rápidamente, puede provocar hemorragias espectaculares (fibrinólisis hemorrágica) intra o postoperatorias (particularmente en Cirugía Torácica), o después de un parto, de un aborto o de un choque traumático; o bien puede provocar hemorragias menos importantes (hematuria) en el curso de ciertos cánceres (próstata, páncreas, estómago), de las cirrosis y de las leucosis. Obedece a la liberación de activadores de la profibrinolisina, o bien a la producción de un fermento proteolítico por los tejidos enfermos.
Plaquetas, coagulación y fribinolisis 1. Activación Plaquetaria: Los procesos de activación plaquetaria comprenden complejas rutas de señalización que involucran sistemas de proteínas G acoplados a enzimas efectoras como la fosfolipasa A2, fosfolipasa C, adenilato y guanilato ciclasas. Las propiedades funcionales y estructurales de las plaquetas están estrechamente relacionadas, lo que les confiere un alto potencial trombogénico, contribuyendo sustancialmente en el desarrollo de un infarto cerebral. Se realizó una revisión de las bases de la agregación, activación, degranulación plaquetaria y su participación en la enfermedad cerebrovascular. Se realizó una actualización del uso de los agentes antiplaquetarios y de la literatura actual referente al uso de bloqueadores de las glicoproteínas adhesivas de membrana. 2. Coagulación de la sangre 1, 2, 3 y 4: La coagulación de la sangre requiere plaquetas también denominadas trombocitos y ciertas proteínas llamadas factores de coagulación. Las plaquetas son células con forma de óvalo que se producen en la médula ósea. La mayoría de los factores de coagulación se generan en el hígado. Cuando los vasos sanguíneos se lesionan, las plaquetas son las primeras en llegar al área de la lesión para sellar la pérdida y reducir o detener temporariamente el sangrado. Pero para que el coágulo sea firme y estable, es necesario que intervengan los factores de coagulación.Los doce factores de coagulación del organismo se enumeran con números romanos la tromboplastina es el factor III. Estos factores actúan juntos en una secuencia especializada, casi como las piezas de un rompecabezas. El coágulo se forma al colocarse la última pieza. Pero si falta una pieza, o ésta es defectuosa, el coágulo no puede formarse. 3. Trombo y Cuagulo: Los coágulos sanguíneos o de fibrina son masas que se forman cuando la sangre se coagula y un trombo es un coágulo de sangre que se forma en un vaso o dentro del corazón y permanece allí. Un émbolo es un trombo que viaja desde el vaso o la cámara del corazón donde se formó a otro lugar del cuerpo y el trastorno causado se llama embolia o embolismo.
4. Fibrinolisis: Es un proceso corporal normal que impide que los coágulos sanguíneo que ocurren en forma natural crezcan y causen problemas. La fibrinólisis primaria se refiere a la descomposición normal de los coágulos. La fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa. Esto puede provocar sangrado intenso.
Av86070 Alejandra Valle: Ya que han abarcado tan bien los temas decidí buscar información relacionada a la agregación plaquetaria y estudios científicos relacionados a ella.
Efecto del ozono sobre la activación plaquetaria en pacientes diabéticos tratados con ozonoterapia: Se verificó in vivo, en los pacientes con diagnóstico de pie diabético isquémico o neuroinfeccioso a los cuales se le indicó tratamiento con 200 mL de ozono, si existía inhibición o no de la agregación plaquetaria al final de este tratamiento (10 a 20 d). La agregación plaquetaria encontrada al inicio, antes del comienzo del tratamiento con ozono, tuvo una media de 28,3 % y un rango de 60 a 6,25 %; mientras que al final del tratamiento con ozono disminuyó a una media de 17,5 % y un rango de 16,25 a 18,75 %. En este estudio preliminar se buscó comprobar si existía o no inhibición del porcentaje de agregación plaquetaria después del tratamiento con ozono, igual a lo observado por otros autores in vitro. Se observó una disminución del porcentaje de agregación plaquetaria al final del tratamiento con ozono, con respecto al valor inicial. Los pacientes incluidos en este estudio tuvieron hiporreactividad plaquetaria, pues no se logró alcanzar en la mayoría 50 % de agregación con concentraciones de epinefrina de hasta 10-5 mol/L, que son suficientes para producir este nivel de agregación en individuos sanos. Esto puede deberse a que, al revisar los tratamientos médicos concomitantes se comprobó, que los pacientes consumían fármacos que no son considerados genéricamente antiagregantes plaquetarios, pero que poseen este efecto.5 Entre estos fármacos se encuentran clorpromazina, defenidramina, fenilbutazona, ibuprofen, imipramina, indometacina, naproxeno propanolol, teofilina, trifluorperazina y otros.
A pesar de la hiporreactividad plaquetaria inicial, el tratamiento con ozono estuvo asociado con una reducción adicional en la agregación plaquetaria determinada en el estudio final.
No se estudiaron los parámetros mediadores del estrés oxidativo, pues en este estudio preliminar se buscaba comprobar si existía o no inhibición del porcentaje de agregación plaquetaria después del tratamiento con ozono, igual a lo observado por otros autores in vitro.
Se observó una disminución del porcentaje de agregación plaquetaria al final del tratamiento con ozono, con respecto al valor inicial.
HEMOSTASIA • La sangre es un tejido líquido que ejerce múltiples funciones esenciales en el organismo • El sistema hemostático es el responsable de mantener a la sangre en estado líquido, prevenir la pérdida de sangre cuando un vaso se rompe y evitar la formación de coágulos indeseables • 2 fases: – Hemostasia primaria: respuesta del vaso y las plaquetas para formar un tapón hemostático primario, inestable – Hemostasia secundaria: formación del coágulo de fibrina. 4 sistemas: Coagulación • Anticoagulación • Fibrinolisis • Antifibrinolisis COAGULACIÓN • Sistema de la coagulación formado por diversas proteínas, casi todas enzimas (síntesis hepática), que circulan de forma inactiva (proenzimas) y que se activan unas a otras secuencialmente formando una cascada de reacciones enzimáticas, tiene lugar sobre una superficie, bien sobre las plaquetas o sobre la membrana de la pared del vaso y requieren la presencia de un cofactor no enzimático y de una superficie fosfolipídica cargada negativamente, que generalmente no está expuesta a la sangre en condiciones normales, lo que hace que se circunscriba el coágulo. • Existen dos vías de inicio de la coagulación: – Vía intrínseca: etapa inicial es la activación del factor XII – Vía extrínseca: se inicia por la exposición de factor tisular al torrente circulatorio. El mecanismo en cascada de la coagulación asegura la rápida y explosiva formación de trombina y del coágulo de fibrina, pero para evitar que todo el árbol vascular se coagule debe existir un mecanismo de control que limite el proceso coagulante • Existen 3 principales mecanismos para este control – El flujo sanguíneo: dispersa la mayor parte de las enzimas generadas, evitando una concentración elevada y permitiendo su inhibición por los inhibidores específicos. – Una superficie cargada negativamente: limita a la zona vascular dañada – Inhibidores específicos de la coagulación
86070 Coagulacion de la sangre: La habilidad del cuerpo para controlar el flujo de sangre luego de una lesión vascular es un componente indispensable de la supervivencia. El proceso de la coagulación sanguínea y luego la disolución del coágulo, seguido por una reparación del tejido lesionado, se denomina hemostasis. La hemostasis se conforma de 4 eventos principales que ocurren en un orden determinado luego de la pérdida de la integridad vascular:
1. La fase inicial del proceso es la constricción vascular. Esto limita el flujo sanguíneo al área de la lesión.
2. A continuación, se activan las plaquetas por la trombina y se agregan en el sitio de la lesión, formando un tampón temporario y flojo conformado de plaquetas. La proteína fibrinógeno es principalmente responsable de estimular la agregación plaquetaria. Las plaquetas se agregan al unirse al colágeno que se expone debido a la ruptura del recubrimiento epitelial de los vasos. Luego de su activación, las plaquetas liberan ADP un nucleótido y un eicosanoide, TXA2 (los cuales activan más plaquetas), serotonina, fosfolípidos, lipoproteínas y otras proteínas importantes de la cascada de coagulación. Además de su secreción, las plaquetas activadas cambian su conformación para acomodar la formación del coágulo.
3. Para asegurar la estabilidad del tampón flojo inicial, se forma una malla de fibrina (también llamada un coágulo) que recubre al tampón. Si el tampón únicamente contiene plaquetas se denomina un trombo blanco; si glóbulos rojos están presentes se lo denomina un trombo rojo
4. Finalmente, el coágulo debe ser disuelto para que el flujo sanguíneo normal pueda resumir luego de que se repare el tejido. La disolución del coágulo ocurre a través de la acción de la plasmina Dos vías llevan a la formación de un coágulo de fibrina: la vía intrínseca y la vía extrínseca. Aunque las dos son iniciadas por mecanismos diferentes, las dos convergen en una vía común que lleva a la formación del coágulo. La formación del trombo rojo ó coágulo en respuesta a una anormalidad en un vaso pero en la ausencia de una lesión del tejido es el resultado de la vía intrínseca. La vía intrínseca tiene poca significancia bajo condiciones fisiológicas normales. El suceso más importante clínicamente es la activación de la vía intrínseca por el contacto de la pared del vaso con las partículas de lipoproteína, VLDLs y quilomicrones. Este proceso claramente demuestra el papel de la hiperlipidemia en la generación de la ateroesclerosis. La vía intrínseca también puede ser activada por el contacto de la pared del vaso con bacterias.
La formación del coágulo de fibrina en respuesta a una lesión del tejido es el evento clínico más importante de la hemostasis bajo condiciones normales de respuesta. Este proceso es el resultado de la activación de la vía extrínseca.
Ambas vías son complejas e incluyen varias proteínas diferentes denominadas factores de la coagulación
El fibrinógeno (factor I) consiste en 3 pares de polipéptidos ([Aα][Bβ][γ])2. Las 6 cadenas están covalentemente unidas cerca de sus N-terminales a través de uniones disulfuro. Las porciones A y B de las cadenas Aα y Bβ constituyen los fibrinopéptidos, A y B, respectivamente. Las regiones de fibrinopéptidos del fibrinógeno contienen varios residuos de glutamato y aspartato impartiendo una alta carga negativa a esta región y promoviendo la solubilidad del fibrinógeno en el plasma. La trombina activa es una proteasa de serina que hidroliza al fibrinógeno en cuatro uniones arg-gly (R-G) entre el fibrinopéptido y las porciones a y b de la proteína.
La liberación de los fibrinopéptidos mediada por la trombina, genera monómeros de fibrina con una estructura de subunidades (αβγ)2. Estos monómeros se agregan espontáneamente en un arreglo regular, formando un coágulo de fibrina relativamente débil. Además de la activación de la fibrina, la trombina convierte el factor XIII al factor XIIIa, una transglutaminasa altamente específica que introduce uniones covalentes entre el nitrógeno de amidas de las glutaminas y el grupo ε-amino de las lisinas pertenecientes a los monómeros de fibrina.
Disolución de los Coágulos de Fibrina
La degradación de los coágulos de fibrina es la función de la plasmina, una proteasa de serina que circula como la proenzima inactiva plasminógeno. Cualquier plasmina libre circulante es rápidamente inhibida por la antiplasmina α2. El plasminógeno se une al fibrinógeno y a la fibrina, y así se incorporan en un coágulo. El activador tisular del plasminógeno (tPA) y, a un grado menor, la urocinasa son proteasas de serina que convierten el plasminógeno a plasmina. El tPA inactivo es liberado de las células endoteliales vasculares luego de una lesión; se une a la fibrina y es consecuentemente activado. La urocinasa es producida como el precursor de la prourocinasa por las células epiteliales que recubren los conductos excretorios. El papel de la urocinasa es activar la disolución de los coágulos de fibrina que pueden depositarse en estos conductos.
La tPA activa cliva al plasminógeno para producir plasmina la cual digiere a la fibrina; esta degradación resulta en un producto soluble al cual ni la plasmina ni el plasminógeno se pueden unir. Luego de la liberación del plasminógeno y la plasmina, éstos se inactivan rápidamente por sus respectivos inhibidores. La inhibición de la actividad de la tPA resulta de la unión de específicas proteínas inhibidoras. Por lo menos 4 diferentes inhibidores han sido identificados, dos de los cuales son inhibidores de los activadores del plasminógeno tipo I (PAI-1) y tipo 2 (PAI-2) tienen la mayor importancia fisiológica.
Los coágulos sanguíneos (coágulos de fibrina) son las masas que se forman cuando la sangre se coagulaLos coágulos sanguíneos o de fibrina son masas que se forman cuando la sangre se coagula y un trombo es un coágulo de sangre que se forma en un vaso o dentro del corazón y permanece allí. Un émbolo es un trombo que viaja desde el vaso o la cámara del corazón donde se formó a otro lugar del cuerpo y el trastorno causado se llama embolia o embolismo (por ejemplo, embolia pulmonar).
Complicaciones Los trombos y émbolos pueden adherirse fírmemente a un vaso sanguíneo y bloquear parcial o completamente el flujo de sangre en dicho vaso. Esta obstrucción priva a los tejidos en esa área del flujo normal de sangre y oxígeno. Esta condición se llama isquemia y, de no tratarse rápidamente, puede ocasionar daño e incluso la muerte de los tejidos (infarto y necrosis) en dicha área.
Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro, derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos. Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva. Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebro vascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores; cualquier anormalidad o enfermedad de las plaquetas es denominada trombocitopatía, la cual puede ser, ya sea un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Hay desórdenes que pueden reducir el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos, pero en cambio otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragia.
Activación
La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP.
Las células endoteliales producen una proteína llamada factor de von Willebrand (FvW), un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas. Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo. Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio.
La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria, estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación. La agregación plaquetaria, usa el fibrinógeno y el FvW como agentes conectores. El receptor de agregación plaquetaria más abundante es la glicoproteina IIb/IIIa (gpIIb/IIIa); se trata de un receptor para el fibrinógeno dependiente del calcio, fibronectina, vitronectina, trombospondina, y factor de von Willebrand (FvW). Otros receptores incluyen el complejo GPIb-V-IX (FvW) y GPVI (colágeno).o Las plaquetas activadas se adherirán, vía glicoproteína (GP) al colágeno expuesto por el daño epitelial. La agregación y adhesión actúan juntos para formar el tapón plaquetario. Los filamentos de Miosina y actina en las plaquetas son estimuladas para contraerse durante la agregación, reforzando todavía más el tapón. La agregación plaquetaria es estimulada por el ADP, tromboxano, y la activación del receptor-α2, pero inhibida por agentes antiinflamatorios como las prostaglandinas PGI2 y PGD2. La agregación plaquetaria se ve aumentada por la administración exógena de esteroides anabólicos. Coagulación sanguínea, cascada de la coagulación.
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre. Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional. El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse. Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas. Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
Factores de coagulación El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc. En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares. A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos. Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas. Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola. Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart
Etapas de la cascada de coagulación, diapositivas 2,3,4,5.
La cascada de coagulación se divide para su estudio, clásicamente en tres vias: La vía intrínseca, la vía extrínseca y la vía común. Las vías intrínseca y extrínseca son las vías de iniciación de la cascada, mientras que la vía común es hacia donde confluyen las otras dos desembocando en la conversión de fibrinógeno en fibrina. Esta división es un tanto arbitraria y tiene más que ver con las deficiencias de las técnicas que en su momento se utilizaron para desentrañar los mecanismos implicados, que con lo que ocurre realmente en una lesión vascular; ya que en este último caso se establecen varias interrelaciones entre las vías de iniciación.
Mecanismo básico Cada reacción de estas vías da como resultado el ensamblado de un complejo compuesto por una enzima (factor de coagulación activado), un sustrato (proenzima de un factor de coagulación) y un cofactor que actúa posibilitando la reacción. Estos componentes se ensamblan en general sobre una superficie fosfolipídica y se mantienen unidos por medio de puentes formados por iones Ca2+. Por lo tanto la reacción en cascada tiende a producirse en un sitio donde este ensamblaje puede ocurrir; por ejemplo sobre la superficie de plaquetas activadas. Tanto la vía intrínseca como la vía extrínseca desembocan en la conversión del factor X en Xa (la letra "a" como subíndice "a" significa "activado") punto en el que se inicia la vía común. Vía intrínseca Recibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso hemostático. El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular. En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación. En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como la celulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción. A esta vía es posible subdividirla en tres pasos: vía intrínseca.
Formación del factor XIa. En esta etapa participan cuatro proteínas: Precalicreína, Quininógeno de alto peso molecular (HMWK) y los factores XII y XI. Esta etapa no requiere de iones calcio. Estos cuatro factores se adsorben sobre la superficie cargada negativamente, formando el complejo cebador o de iniciación. De estos factores el XII funciona como verdadero iniciador, ya que si bien es una proenzima, posee una pequeña actividad catalítica que alcanza para activar a la precalicreína convirtiéndola en calicreína. En segunda instancia la calicreína actúa catalíticamente sobre el factor XII para convertirlo en XIIa, una enzima muchísimo más activa. La actividad catalítica de la calicreína se ve potenciada por el HMWK. Por último la proteasa XIIa actúa sobre el factor XI para liberar XIa.
El factor IX se encuentra en el plasma como una proenzima. En presencia de iones Ca2+ el factor XIa cataliza la ruptura de una unión peptídica en la molécula del factor IX para formar un glucopéptido de 10 KDa y liberar por otro lado al factor IXa. El factor IX se encuentra ausente en personas con hemofilia tipo B.
Formación del factor Xa Sobre la membrana de las plaquetas se forma un complejo constituido por los factores IXa, X y VIII, Los residuos gamma-carboxiglutamato de los factores IXa y X actúan como quelantes del ion Ca2+, permitiendo que estos componentes formen un complejo unido por medio de puentes de iones calcio y ayudando a que el complejo se ancle a los fosfolípidos de membrana. Primero se unen los factores X y IXa a la membrana y luego se une el VIII. El factor VIII es en realidad un homorodímero, formado por cuatro cadenas proteicas, cada una codificada por un gen diferente (VIII:C y VIII:R). El componente VIII: C es conocido como "componente antihemofílico" y actúa como cofactor del IXa en la activación del factor X, el componente VIII: R es el que permite la unión del factor VIII al complejo. La ausencia del componente antihemofílico causa hemofilia A. El complejo formado por los factores IXa-X-VIII-Fosfolípidos y Ca2+ actúa sobre el factor X para convertirlo en Xa. En este punto concluye la vía intrínseca.
Vía extrínseca. Recibió este nombre debido a que fue posible notar desde un primer momento que la iniciación de esta vía requería de factores ajenos a la sangre. Cuando la sangre entra en contacto con tejidos lesionados o se mezcla con extractos de tejidos, se genera muy rápidamente factor Xa. En este caso la activación de la proenzima X es mediada por un complejo formado por factor VII, Ca2+ y factor tisular unido a fosfolípidos provenientes de las membranas celulares rotas y de las plaquetas (antiguamente este complejo factor tisular-fosfolípidos era conocido como tromboplastina). El factor tisular es una lipoproteína sintetizada en el endotelio de los vasos sanguíneos de todos los tejidos, aunque es especialmente abundante en pulmón, cerebro y placenta. El factor tisular se encuentra normalmente "secuestrado" en el interior de las células endoteliales y es secretado en respuesta a una lesión, o bajo el efecto de algunas citoquinas tales como el Factor de Necrosis Tumoral (TNF), Interleucina 1 (IL-1); o por endotoxinas bacterianas. La vía extrínseca es muy rápida, se cumple en apenas unos segundos y comprende dos pasos; mientras que la intrínseca insume varios minutos.
Formación del factor VIIa En primera instancia el factor VII se une a la porción fosfolipídica del factor tisular gracias a sus residuos gamma-carboxiglutamato, utilizando iones Ca2+ como puentes. Este complejo provoca la activación del factor VIIa.
El complejo VIIa-III-Ca2+ actúa sobre el factor X convirtiéndolo en la proteasa activa Xa. En este punto termina la vía extrínseca y se inicia la vía común.
Vía común Llegando al punto en que se activa el factor X, ambas vías confluyen en la llamada vía común. La vía común termina con la conversión de fibrinógeno en fibrina, y el posterior entrecruzamiento de la misma estabilizando el coágulo. La vía común implica tres etapas:
Formación de trombina La trombina (también llamada factor II a) es una proteasa generada por la ruptura de la cadena proteica de la proenzima protrombina (factor II), una glicoproteína constituida por 582 aminoácidos y con 12 puentes disulfuro intracatenarios. La trombina se activa luego de que la proteasa Xa hidroliza dos uniones peptídicas de la protrombina. La Xa produce en primer término la escisión de un fragmento de 32 KDa de la región N-terminal de la cadena, cortándola sobre una unión arginina-treonina. En segundo término produce la ruptura de un enlace entre una arginina y una isoleucina; sin embargo estos dos últimos fragmentos permanecen unidos por un puente disulfuro. La trombina es una serina-proteasa similar a la tripsina, pero mucho más selectiva. Ataca casi de manera exclusiva las uniones arginina con un aminoácido cargado positivamente en sus sustratos. La conversión de protrombina a trombina debida al factor Xa se acelera notablemente por la formación de un complejo con el factor Va y Ca2+ sobre la superficie de las membranas plaquetarias (fosfolípidos de membrana). El factor Xa y la protrombina se adsorben sobre la membrana utilizando iones Ca2+ como puentes. El factor Va se une a la protrombina acelerando la reacción. El factor Va se produce por la acción de la trombina sobre el factor V en un claro ejemplo de una reacción que va acelerándose a medida que progresa (reacción autoacelerada).
Formación de fibrina El fibrinógeno (factor I) es una glicoproteína compuesta por seis cadenas polipeptídicas: dos A-alfa, dos B-beta y dos gamma; unidas entre sí por puentes disulfuro. Se trata de una molécula alargada y simétrica formada por tres dominios globulares conectados por segmentos fibrilares. Cada mitad de la molécula se encuentra formada por tres cadenas (A-alfa, B-beta y gamma) que se enrollan en una triple hélice muy compacta en los sectores fibrilares. Los extremos amino de las seis cadenas se reúnen en el dominio globular central. En un hecho que parecería muy curioso, los extremos N-terminales de las cadenas A-alfa y B-beta emergen como cabos libres del dominio globular central.
Representación de la molécula de fibrinógeno y cómo, al eliminarse los fibrinopéptidos, polimeriza para formar un agregado de fibrina. Estas cadenas son muy ricas en aspartato y glutamato, además las cadenas B-beta poseen en esta región residuos tirosina-O-sulfato formados postraduccionalmente. Estos residuos con una alta tendencia a adquirir carga negativa contribuyen a formar una región central con una muy alta densidad de carga. Esta región electronegativa central es la responsable de la repulsión entre moléculas de fibrina que las mantiene en solución. La trombina ataca los enlaces arginina-glicina presentes en estos "cabos libres", separando cuatro péptidos; dos segmentos A de 18 aminoácidos cada uno (provenientes de las cadenas A-alfa), y dos segmentos B de 20 aminoácidos (provenientes de las cadenas B-beta). A estos péptidos se los suele denominar "fibrinopéptidos".El resto que queda de la molécula es un monómero de fibrina de composición alfa2beta2gamma. Al eliminarse los fibinopéptidos desaparecen las fuerzas de repulsión intermoleculares con lo que los monómeros de fibrina tienden a agruparse espontáneamente formando asociaciones altamente ordenadas. Entrecruzamiento de la fibrina Los haces paralelos de fibrina polimerizada forman una asociación laxa, que se encuentra en equilibrio con la forma monomérica de la molécula; por lo que sería imposible que cumplieran su papel de formar un coágulo estable sin reforzar esta estructura por medio de enlaces covalentes entre hebras vecinas. La formación de estos "puentes" covalentes intercatenarios es catalizada por la enzima transglutaminasa (conocida también como factor XIIIa). La transglutaminidasa cataliza la formación de enlaces amida entre restos glutamina y lisina de hebras próximas entre sí. En la reacción se libera amoníaco en forma de ion amonio (NH4+). Esta enzima se forma a partir del factor XIII por acción de la trombina. Regulación y modulación de la cascada (de forma general). Debido a que la cascada de coagulación consiste en una serie de reacciones que van amplificándose y acelerándose en cada paso, es lógico pensar que debe existir algún mecanismo de regulación; un "freno" a la reacción en cadena; ya que de progresar sin control en pocos minutos podría provocar un taponamiento masivo de los vasos sanguíneos (trombosis diseminada). Varios mecanismos intervienen en la regulación de la cascada de reacciones: El flujo sanguíneo normal, arrastra a los factores activados, diluyendo su acción e impidiéndoles acelerarse. Esta es una de las razones por las cuales cuando existe estasis del flujo sanguíneo se favorece la formación de trombos. El hígado actúa como un filtro quitando de la sangre en circulación los factores activados e inactivándolos. Existen además algunas proteasas que degradan específicamente a ciertos factores activados, y otras que ejercen acciones inhibitorias sobre factores activos.
La proteína C es una proenzima que se encuentra normalmente en el plasma, y cuya síntesis en el hígado es dependiente de la vitamina K. Esta proteína es convertida en una proteasa activa por la acción de la trombina. La proteína Ca actúa específicamente degradando a los factores Va y VIIIa, con lo que limita la proyección de la cascada. Es interesante notar el triple papel que desempeña la trombina: cataliza la formación de fibrina, activa a la enzima responsable de su entrecruzamiento, y una vez que el proceso de coagulación y estabilización del coágulo está en marcha; ejerce acciones tendientes a limitarlo.
Antitrombina III La antitrombina III es una glicoproteína de 60 kDa sintetizada en el hígado sin depender de la vitamina K, es considerada la principal inhibidora de la coagulación. Esta proteína actúa inhibiendo irreversiblemente a varios factores procoagulantes activos, el principal de los cuales es la trombina; aunque también actúa sobre la calicreína y los factores IXa, Xa, XIa y XIIa. La acción de la antitrombina es notablemente aumentada por el heteropolisacárido heparina. La heparina se encuentra en el endotelio de los vasos sanguíneos y en los gránulos de las células cebadas, tiene una poderosa acción anticoagulante ya que facilita la unión de la antitrombina III con los factores procoagulantes activos. Existen otras anti-proteasas sanguíneas que también ejercen acción anticoagulante aunque menos potente tales como la alfa2 macroglobulina y la alfa1 antitripsina.
La trombosis es un coágulo en el interior de un vaso sanguíneo y uno de los causantes de un infarto agudo de miocardio. También se denomina así al propio proceso patológico, en el cual, un agregado de plaquetas o fibrina ocluye un vaso sanguíneo. Cuando un vaso sanguíneo se lesiona, el cuerpo utiliza plaquetas (trombocitos) y fibrina para formar un coágulo de sangre para prevenir la pérdida de sangre. Incluso cuando un vaso sanguíneo no se lesione, los coágulos de sangre se pueden formar en el cuerpo en ciertas condiciones. Un coágulo que se desprende y comienza a viajar por todo el cuerpo se conoce como embolia. El trombo es una masa que se forma en el interior del aparato circulatorio y está constituida por la sangre del paciente, específicamente por los elementos sólidos de la sangre. En un individuo vivo la sangre puede coagularse pero esto es fuera del aparato circulatorio por ejemplo, la sangre que pasa al peritoneo, a la pleura o al pericardio. Ahí la sangre coagula y no es un trombo, porque está fuera del aparato cardiovascular.
Las causas son: Alteración en los vasos sanguíneos: Arteriosclerosis. ruptura traumática. Alteración en los factores de la coagulación: trombina, protrombina. disminución de la Proteína C, Proteína S, llamadas estas últimas trombofilias. Los mecanismos que favorecen la formación de un trombo, son las alteraciones del flujo sanguíneo y estas alteraciones pueden deberse a reposo excesivo en cama (pacientes postoperados). Además en la intervención quirúrgica ha habido una estimulación de los factores de coagulación por la rotura de vasos, la sutura, una serie de intervenciones que involucran al aparato vascular. No es raro que un individuo se opere de una hernia inguinal, y en el momento que se le da de alta y empieza a moverse más de lo que se ha movido en los días anteriores presente una embolia fulminante ocasionándole la muerte. La tercera causa que influye son los componentes de la sangre. Cuando la sangre es más densa disminuyen los líquidos y aumentan los elementos figurados. O hay una hemoconcentración o una policitemia real. Dentro de este se incluye las trombosis a repetición.
Los sitios de formación de trombo son en el corazón, arterias, venas y capilares, por lo que la trombosis puede formarse en cualquier parte del aparato circulatorio. Otras patologías que pueden provocar una trombosis son aquellas que presentan flujos en torbellinos, como las estrecheces valvulares. Un ejemplo es la estenosis mitral, en donde el flujo en la aurícula se hace más lento y favorece la trombosis. En la estenosis mitral hay que tener en cuenta que lo más probable es que haya trombosis en la orejuela y en alguna parte de la pared de la aurícula. Y si ésa hace flutter o fibrilación, la contracción de la aurícula es ineficiente. Entonces la aurícula no se contrae y además existe oclusión en la salida, de tal modo que hay una lentitud del flujo de salida y por tanto formación de coágulos (trombos). Otra causa de trombosis es el daño del endotelio. Si un vaso se inflama, por ejemplo una vena de las EEII por un trauma, se produce una lesión de la vecindad y daño endotelial, que desencadena inmediatamente la cascada de coagulación depositándose trombos en la superficie del vaso.
Factores de riesgos Hay tres tipos de factores de riesgo:
Primarios Congénitos: Falta de la proteína C, que evita la coagulación. Resistencia a la proteína C activada. Adquiridos: Anticuerpos antifosfolípido. Secundarios Anomalías en las plaquetas. Anomalías vasculares. Exceso de tabaco. Exceso de arsénico (elemento utilizado en el veneno de ratas, por ejemplo).
Las trombosis pueden clasificarse según el nivel de oclusión que alcanzan y el lugar en el que se originan.-Según el grado de oclusión, podemos diferenciar entre trombos ocluyentes y murales. Los primeros son aquellos en los que el vaso queda completamente obstruido por la afección, mientras que en los murales, el resultado es una obstrucción parcial. Dependiendo de la ubicación, las trombosis se clasifican según tres tipos: por precipitación, hialina o por coagulación.
Trombosis por precipitación (Trombos Blancos): producida principalmente en arterias o el corazón, se deben al desprendimiento de plaquetas. Son de carácter mural. Trombosis hialina: producida en vénulas o capilares. Suelen ser provocadas por el desprendimiento de plaquetas y fibrinas. Trombosis por coagulación (Trombos Rojos): producida en las venas suelen ser de naturaleza oclusiva y deberse a una mezcla de plaquetas y fibrinas, apareciendo estas últimas en una mayor proporción que las primeras. Esta situación es de extrema gravedad, pues el territorio más allá del trombo deja de recibir irrigación sanguínea, produciéndose inicialmente isquemia y luego muerte de las estructuras. Se puede producir la parálisis de los músculos si se encuentran en el trombo que se ubica en una vena. Dependiendo de la ubicación de la vena, estas trombosis pueden ser graves (trombosis del seno cavernoso), de mediana gravedad (trombosis venosa profunda) o leves (tromboflebitis superficial).
Diferencia entre Trombosis y Embolia Una trombosis es la obstrucción de un vaso sanguíneo que, generalmente, es producida por una placa de arteriosclerosis que crece en la pared de dicho vaso. Si la placa se desprende, se denomina émbolo y puede ir hacia diversos lugares del organismo, en mayor medida al pulmón. Pero los émbolos pueden ser de material diverso, no solamente trombos, también émbolos infecciosos, de cristales de colesterol, etc. No confundir con la Trombosis hemorroidal.
Tratamiento. La prevención de coágulos de sangre y el tratamiento reduce los riesgos de ataque al corazón, accidente cerebrovascular y embolia pulmonar. La heparina y warfarina se utilizan a menudo para inhibir la formación y crecimiento de trombos existentes, el primero se une y activa la enzima inhibidor antitrombina III, mientras que el segundo inhibe la vitamina K epóxido reductasa, una enzima necesaria para sintetizar maduros factores de coagulación.
Coágulos. Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida.
Causas Las situaciones en las que es más probable que se forme un coágulo de sangre abarcan: Estar en reposo en cama por largo tiempo. Cruzar las piernas durante largos periodos al estar sentado o sentarse durante mucho tiempo, como en un avión o en un vehículo. Durante y después del embarazo. No tener suficiente agua en el cuerpo (deshidratación). Tomar píldoras anticonceptivas u hormonas estrógeno (especialmente en las mujeres que fuman). Utilizar un catéter intravenoso por largo tiempo. Los coágulos de sangre también son más probables en personas con cáncer, cirugía o lesión reciente, obesidad y enfermedades del hígado o del riñón. Una acumulación de colesterol que estreche una arteria puede cambiar o reducir el flujo de sangre, facilitando la formación de un coágulo sanguíneo o de un trombo. Las afecciones que se transmiten de padres a hijos (hereditarias) pueden hacerlo más propenso a la formación de coágulos sanguíneos anormales.
Los trastornos hereditarios que afectan la coagulación son: Trombofilia por factor V de Leiden Mutación de la protrombina G20210A Otras afecciones raras como deficiencias de antitrombina III, proteína C y S
Un coágulo de sangre puede bloquear una arteria o una vena en el corazón, afectando: El corazón (angina de pecho o un ataque cardíaco). Los intestinos (isquemia mesentérica) o trombosis venosa mesentérica. Los riñones (trombosis de la vena renal). Las arterias de las piernas o brazos. Las piernas (trombosis venosa profunda). Los pulmones (embolia pulmonar). El cuello o el cerebro (accidente cerebrovascular). Posibles complicaciones Los trombos y émbolos pueden fijarse firmemente a un vaso sanguíneo y pueden bloquear parcial o completamente el flujo de sangre a dicho vaso.Un bloqueo en el vaso sanguíneo impide que el flujo normal de sangre y oxígeno llegue a los tejidos en ese sitio, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede ocasionar daño tisular o necrosis en esa área.
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos.
Bioquímica La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinólisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón.
La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca: En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA.
En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno.
Regulación La fibrinólisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Consecuencias Cuando se produce demasiado rápidamente, puede provocar hemorragias espectaculares (fibrinólisis hemorrágica) intra o postoperatorias (particularmente en Cirugía Torácica), o después de un parto, de un aborto o de un choque traumático; o bien puede provocar hemorragias menos importantes (hematuria) en el curso de ciertos cánceres (próstata, páncreas, estómago), de las cirrosis y de las leucosis. Obedece a la liberación de activadores de la profibrinolisina, o bien a la producción de un fermento proteolítico por los tejidos enfermos.
Tipos Puede ser primitiva aunque por lo general es secundaria, reactiva, en el curso de un síndrome de coagulación intravascular diseminada Causas, incidencia y factores de riesgo Los coágulos de sangre se forman a partir de una proteína llamada fibrina. La descomposición de la fibrina (fibrinólisis) puede incrementarse bajo ciertas condiciones, tales como: Infecciones bacterianas Ejercicio intenso Glucemia baja Oxigenación insuficiente a los tejidos Procedimiento médico alternativo
En algunas situaciones, es posible que los médicos quieran acelerar el proceso de fibrinólisis. Por ejemplo, cuando se forma un coágulo sanguíneo anormal en los vasos sanguíneos del corazón y provoca un ataque cardíaco, se pueden suministrar sustancias fibrinolíticas sintéticas (como, estreptocinasa o Retavase) para disolverlo.
El sistema fibrinolítico va a jugar además un papel importante en diversas situaciones en las que se produce proteolisis tisular, tales como inflamación, invasión tumoral o neovascularización. La importancia fisiopatológica del sistema fibrinolítico deriva de que las alteraciones que condicionan un defecto de actividad fibrinolíticas pueden predisponer a la trombosis, mientras que un exceso de activación favorecería la aparición de hemorragia. Además va a tener una gran importancia en el área terapéutica por la aplicación creciente de fármacos fibrinolíticos en el tratamiento de la trombosis, especialmente en el infarto agudo de miocardio. Principales componentes del sistema fibrinolítico: La fibrinólisis es un encadenamiento (proceso) enzimático compuesto por una serie de activadores e inhibidores los cuales regulan la conversión del proenzima circulante, plasminógeno, en el enzima activo plasmina.
La producción de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina y esto es muy importante para el mantenimiento de la permeabilidad vascular. Los principales componentes del sistema fibrinolítico son los siguientes:
PLASMINÓGENO Es una glicoproteína monocatenaria compuesta por 790 aminoácidos y 24 puentes disulfuro con un contenido en carbohidratos del 2% y un peso molecular (Pm) de 92.000 daltons, sintetizándose en el hígado; su concentración plasmática es de 2mM (20 mg/dl) y su vida media de 2,2 días. El gen que codifica esta proteína está localizado en el cromosoma 6. En su forma nativa el plasminógeno contiene ácido glutámico en posición amino-terminal (Glu-plasminógeno), pudiendo ser convertido por pequeñas cantidades de plasmina a formas con lisina, valina o metionina en dicha posición, denominadas Lis-plasminógeno, los cuales poseen mayor afinidad por la fibrina y se activan más fácilmente por los activadores fibrinolíticos.
ACTIVADORES DEL PLASMINÓGENO La activación del plasminógeno a plasmina tiene lugar por escisión del enlace Arg561-Val562 en la molécula del Glu-plasminógeno por acción de los diferentes activadores. La plasmina así formada es una serín-proteasa bicatenaria compuesta por una cadena pesada derivada de la porción amino-terminal, que contiene 560 aminoácidos y 5 “kringles” en donde se localizan los LBS, y una cadena ligera derivada de la posición carboxiterminal compuesta por 241 aminoácidos, que contiene el centro activo formado por los aminoácidos serina, histidina y ácido aspártico. En la actualidad se distinguen varias vías de activación.
Activación intrínseca del plasminógeno Fue denominada inicialmente activación por contacto o XII-dependiente por estar implicados diversos factores del sistema de contacto de la coagulación. Actualmente se sabe que además del factor XII existen otras sustancias en la sangre, denominadas precursores, como la precalicreína o el quininógeno de alto peso molecular que pueden inducir la activación del plasminógeno. La activación del factor XII conduce a la generación de calicreína que va a ser un potente activador de la fibrinólisis intrínseca.
Activador tisular del plasminógeno (t-PA) Es una proteasa serínica monocatenaria constituida por 530 aminoácidos y un Pm de 68.000 daltons la cual puede ser convertida en una molécula de doble cadena por acción de la plasmina. La cadena pesada A (1-278aa) contiene dominios homólogos a los encontrados en otras proteínas: una región homóloga a la de la fibronectina, denominada dominio “finger”, una estructura similar al factor de crecimiento epidérmico y dos “kringles” similares a los del plasminógeno.
Activadores tipo uroquinasa y prouroquinasa. La UK es una proteasa serínica similar a la tripsina, compuesta por dos cadenas polipeptídicas de 20.000 y 34.000 daltons unidas por un puente disulfuro; fue aislada inicialmente a partir de orina humana y cultivo de células embrionarias de riñón humano. Es un activador directo del plasminógeno mediante escisión de un enlace Arg561-Val562 con formación de Glu-plasmina pero, a diferencia del t-PA, carece de afinidad específica por la fibrina, ya que activa indiscriminadamente tanto al plasminógeno circulante como al unido a la fibrina. Tanto su mecanismo de acción como sus características farmacocinéticas serán revisadas en profundidad más adelante.
La prouroquinasa es una glicoproteína monocatenaria compuesta por 411 aminoácidos y un Pm de 54.000 daltons. La porción amino-terminal contiene una región homóloga al factor de crecimiento epidérmico y otra similar a los “kringles” del plasminógeno, pero a diferencia del t-PA no posee un dominio “finger, lo que quizás pudiera explicar su falta de afinidad por la fibrina. En la región carboxi-terminal se encuentra su centro activo el cual contiene el aminoácido serina.
Activación exógena del plasminógeno. Consiste en la activación que se produce por la acción de diferentes sustancias exógenas administradas con fines terapéuticos.
INHIBIDORES DEL SISTEMA FIBRINOLÍTICO Se clasifican en inhibidores competitivos del plasminógeno, inhibidores de los activadores del plasminógeno e inhibidores de la plasmina. MODULADORES DE LA ACTIVIDAD FIBRINOLÍTICA. La actividad fibrinolítica plasmática viene determinada por un balance entre los activadores e inhibidores, fundamentalmente t-PA y PAI-1. Ambas sustancias están sometidas a una estrecha regulación por la acción de diversos estímulos fisiológicos y patológicos. Así, por ejemplo, el estrés, ejercicio físico, oclusión venosa o administración de fármacos vasoactivos aumentan la actividad fibrinolítica plasmática al favorecer la liberación de t-PA desde el endotelio vascular. La insulina aumenta la síntesis de PAI-1 por los hepatocitos, aunque no tiene efecto sobre las células endoteliales. Los corticoides y especialmente la dexametasona, disminuyen la liberación de t-PA e inducen la síntesis de PAI-1. Diversas citoquinas como la interleuquina 1 (IL-1) y el factor de necrosis tumoral (TNF) inducen una disminución de t-PA y un aumento de PAI-1. Finalmente, la endotoxina, ya sea directamente o a través de citoquinas, inducen un marcado aumento del PAI-1 por el endotelio vascular.
Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro, derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos. Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva. Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebro vascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores. Cualquier anormalidad o enfermedad de las plaquetas se denomina trombocitopatía,2 la cual puede consistir, ya sea en tener un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Se pueden producir desórdenes que reducen el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos. Sin embargo, otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragias.
Las plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular; Estos dos factores de crecimiento han demostrado jugar un papel significativo en la regeneración y reparación del tejido conectivo; Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.
Cinética Las plaquetas son producidas en el proceso de formación de las células sanguíneas llamado (trombopoyesis) en la médula ósea, por fragmentación en los bordes citoplasmáticos de los megacariocitos. El rango fisiológico de las plaquetas es de 150-400 x 109/litro. Un adulto sano produce cada día alrededor de 1 x 1011 plaquetas de media. La expectativa de vida de las plaquetas circulantes es de 7 a 10 días. La producción de megacariocitos y plaquetas está regulada por la trombopoyetina, una hormona producida habitualmente por el hígado y los riñones. Cada megacariocito produce entre 5.000 y 10.000 plaquetas, Las plaquetas son destruidas por fagocitosis en el bazo y por las células de Kupffer en el hígado. Una reserva de plaquetas es almacenada en el bazo y son liberadas cuando se necesitan por medio de contracción esplénica mediada por el sistema nervioso simpático. Formación de trombos. La función plaquetaria consiste en el mantenimiento del corazón; Esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio. Activación. La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP. Las células endoteliales producen una proteína llamada factor de von Willebrand (FvW),un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas. Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo. Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio. La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria (plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
Cambio de forma. Las plaquetas activadas cambian su forma haciéndose más esféricas, y formando pseudopodos en su superficie. De esta forma toman una forma estrellada.
Secreción de gránulos. Las plaquetas contienen gránulos alfa y gránulos densos. Las plaquetas activadas excretan el contenido de estos gránulos dentro de sus sistemas canaliculares y en la sangre circundante. Existen dos tipos de gránulos: Gránulos densos (contienen ADP o ATP, calcio, y serotonina). Granulos-α (contienen factor 4 plaquetario, factor de crecimiento transformante beta 1 (TGF beta 1), factor de crecimiento derivado de plaquetas, fibronectina, B-tromboglobulina, FvW, fibrinógeno, y factores de coagulación factor V y XIII).
Síntesis de tromboxano A2. La activación plaquetaria inicia la vía del ácido araquidónico para producir Tromboxano A2; el tromboxano A2 está involucrado en la activación de otras plaquetas y su formación es inhibida por los inhibidores de la COX, como el ácido acetilsalicílico.
Adhesión y agregación. La agregación plaquetaria, usa el fibrinógeno y el FvW como agentes conectores. El receptor de agregación plaquetaria más abundante es la glicoproteina IIb/IIIa (gpIIb/IIIa); se trata de un receptor para el fibrinógeno dependiente del calcio, fibronectina, vitronectina, trombospondina, y factor de von Willebrand (FvW). Otros receptores incluyen el complejo GPIb-V-IX (FvW) y GPVI (colágeno).ó Las plaquetas activadas se adherirán, via glicoproteína (GP) Ia, al colágeno expuesto por el daño epitelial. La agregación y adhesión actúan juntos para formar el tapón plaquetario. Los filamentos de Miosina y actina en las plaquetas son estimuladas para contraerse durante la agregación, reforzando todavía más el tapón.
La agregación plaquetaria es estimulada por el ADP, tromboxano, y la activación del receptor-α2, pero inhibida por agentes antiinflamatorios como las prostaglandinas PGI2 y PGD2. La agregación plaquetaria se ve aumentada por la administración exógena de esteroídes anabólicos.
Reparación de heridas. El coágulo sanguíneo es solo una solución temporal para detener la hemorragia; la reparación del vaso debe ocurrir después. La agregación plaquetaria ayuda en este proceso mediante la secreción de sustancias químicas que promueven la invasión de fibroblastos del tejido conectivo adyacente hacia el interior de la herida para formar una costra. El coágulo obturador es lentamente disuelto por la enzima fibrinolítica, plasmina, y las plaquetas son eliminadas por fagocitosis.
Otras funciones Retracción del coágulo Pro-coagulación Inflamación Señalización citoquínica Fagocitosis
Señalización citoquínica Adicionalmente a su función de ser el efector celular de la hemostasia, las plaquetas son rápidamente depositadas en sitios de lesión o infección, y potencialmente modulan los procesos inflamatorios por medio de la interacción con leucocitos y por la secreción de citoquinas, quimiosinas, y otros mediadores de la inflamación las plaquetas también secretan factor de crecimiento derivado de plaquetas (PDGF). Papel en enfermedades. Recuentos altos y bajos. El recuento de plaquetas de un individuo sano se encuentra entre 150,000 y 450,000 por μl (microlitro) de sangre (150-450 x 109/L).18 El 95 % de los individuos sanos tendrán recuentos de plaquetas dentro de este rango. Algunos tendrán recuentos de plaquetas estadísticamente anormales sin tener ninguna anormalidad demostrable. Sin embargo, si el recuento es muy alto o muy bajo la probabilidad de que una anormalidad este presente es más alta. Tanto la trombocitopenia como la trombocitosis pueden manifestarse como problemas de coagulación. En general, los recuentos bajos de plaquetas incrementan el riesgo de sangrado; sin embargo existen excepciones. Por ejemplo la trombocitopenia inmune inducida por heparina. En la trombocitosis se puede producir trombosis, sin embargo esto sucede principalmente cuando el recuento elevado es debido a desordenes mieloproliferativos. Los recuentos de plaquetas en general, no son corregidos con transfusión a menos que el paciente esté sangrando o el recuento haya caído por debajo 5 x 109/L. La transfusión está contraindicada en la púrpura trombocitopénica idiopática (PTI), puesto que estimula la coagulopatía. En los pacientes sometidos a cirugía, niveles inferiores a 50 x 109/L están asociados a sangrado quirúrgico anormal, y procedimientos anestésicos regionales como la anestesia epidural son evitados para niveles inferiores a 80-100 x 109/L. El recuento normal de plaquetas no es garantía de función adecuada. En algunos estados, las plaquetas, siendo normales en número, son disfuncionales. Por ejemplo, el ácido acetilsalicílico interrumpe irreversiblemente la función plaquetaria mediante la inhibición de la ciclooxigenasa-1 (COX1), y por consiguiente la hemostasia normal. Las plaquetas resultantes no tienen ADN y son incapaces de producir nueva ciclooxigenasa. La función plaquetaria normal no se restaurara hasta que el uso de ASA haya cesado y un número suficiente de las plaquetas afectadas hayan sido reemplazadas por nuevas, lo cual suele tardar unos 7 días. El Ibuprofeno, un AINE, no tiene un período tan largo de efecto, y la función plaquetaria vuelve a la normalidad dentro de las 24 horas, y tomando ibuprofeno antes que el ASA prevendrá los efectos irreversibles de esta. La Uremia, a consecuencia de la insuficiencia renal, conduce a la disfunción plaquetaria que puede ser aminorada con la administración de desmopresina.
Medicamentos • Agentes orales usados a menudo para alterar/suprimir la función plaquetaria: • Ácido acetilsalicílico • Clopidogrel • Cilostazol • Ticlopidina
Agentes intravenosos usados a menudo para alterar/suprimir la función plaquetaria: • Abciximab • Eptifibatida • Tirofiban
Enfermedades. Desordenes que provocan un recuento bajo de plaquetas: • Trombocitopenia • Púrpura trombocitopénica idiopática • Púrpura trombocitopénica trombótica • Púrpura trombocitopenica inducida por medicamentos • Enfermedad de Gaucher • Anemia aplásica • Trastornos Aloimunes • Trombocitopenia fetomaterna autoinmune • Algunas reacciones trasfusionales
Desordenes que provocan disfunción o recuento reducido: • Síndrome HELLP • Síndrome urémico hemolítico • Quimioterapia • Dengue • Deficiencia del almacenamiento en gránulos Alfa-Delta; es un desorden hemorrágico hereditario.
Desordenes caracterizados por recuentos elevados: • Trombocitosis, incluyendo trombocitosis esencial (recuento elevado, ya sea reactivo o como una expresión de trastorno mieloproliferativo); puede mostrar plaquetas disfuncionales. • Desordenes de la agregación y adherencia plaquetarios: • Síndrome de Bernard-Soulier • Tromboastenia de Glanzmann • Síndrome de Scott's • Enfermedad de von Willebrand • Síndrome de Hermansky-Pudlak • Síndrome de plaquetas grises • Desordenes del metabolismo de plaquetas • Actividad disminuida de la oxigenación, inducida o congénita • Defectos del almacenamiento, adquirido o congénito
Desordenes que comprometen la función plaquetaria: • Hemofilia • Desordenes en los cuales las plaquetas juegan un papel clave: • Aterosclerosis • Enfermedad coronaria arterial, e infarto del miocardio • Accidente cerebro vascular • Enfermedad arterial oclusiva periférica • Cancer
La preparación del plasma rico en plaquetas (PRP) de procedencia autóloga, requiere la extracción y recolección de sangre periférica del paciente, la separación de las plaquetas y el plasma de los otros elementos formes sanguíneos, y la posterior polimerización de la fibrina de dicho plasma para concentrar las plaquetas formando un gel rico en plaquetas con suficiente estabilidad como para ser implantado quirúrgicamente. Actualmente, algunos métodos comerciales para la preparación del PRP utilizan calcio y trombina bovina o bien, trombina preparada de forma autóloga para crear una matriz rica en plaquetas y fibrina. La preparación de trombina autóloga requiere tiempo y pasos adicionales, así como un mayor volumen de sangre; por otro lado, el uso de trombina bovina ha sido asociado con el desarrollo de anticuerpos contra los factores de coagulación V y XI, y la misma trombina, aumentando de esta forma el riesgo de anormalidades en la coagulación. Adicionalmente, para asegurar una degranulación de las plaquetas y la formación de un coágulo estable, se utilizan grandes cantidades de trombina, esto puede causar una liberación inmediata de los factores de crecimiento.
La liberación de factores de crecimiento es desencadenada por la activación de las plaquetas, esta puede ser iniciada por una gran variedad de sustancias o estímulos como la trombina, el cloruro de calcio, el colágeno o el adenosina 5c-difosfato. Un coágulo sanguíneo de PRP contiene aproximadamente un 4% de glóbulos rojos, 95% de plaquetas y 1% de glóbulos blancos. Las propiedades del PRP están basadas en los múltiples factores de crecimiento y de diferenciación producidos y liberados a raíz de la activación de las plaquetas. Estos factores son críticos para la regulación y estimulación del proceso curativo de lesiones. Por otro lado, existe una segunda generación del concentrado de plaquetas que recibe el nombre de matriz rica en plaquetas y fibrina la cual es un mejoramiento del PRP preparado tradicionalmente. Existe un método actual que evita el uso de trombina como activador Este sistema utiliza únicamente calcio y centrifugación para activar la polimerización de la fibrina y formar así el PRFM. PRFM, en forma de gel o una membrana densa y flexible, puede ser aplicada al paciente y la liberación de los factores de crecimiento es desencadenada por los activadores autólogos presentes en el sitio de aplicación. Este método permite una liberación gradual de los factores de crecimiento en el sitio de aplicación, que pueden emitir señales a diferentes tipos celulares para que emitan una respuesta en momentos apropiados. Estudios in vitro indican que el PRFM presenta una liberación gradual y estable de los factores de crecimiento a lo largo de 7 días. El plasma rico en plaquetas (PRP) puede obtenerse por medio de diferentes técnicas ya sean separadores celulares de propósitos generales o bien, separadores celulares para la concentración de plaquetas. Muchos productos comerciales se encuentran disponibles en este campo, la mayoría de ellos obtienen resultados similares, cuyas diferencias se deben fundamentalmente al precio, tiempo, espacio requerido y la tecnología necesaria para fabricarlo. Son pocos los productos comerciales disponibles para la obtención de una matriz rica en plaquetas y fibrina como producto final.
stephanie de los santos 86182 Plaquetas Las plaquetas son pequeñas células que circulan en la sangre; participan en la formación de coágulos sanguíneos y en la reparación de vasos sanguíneos dañados. Cuando un vaso sanguíneo se lesiona, las plaquetas se adhieren al área dañada y se distribuyen a lo largo de la superficie para detener la hemorragia (este proceso se conoce como adhesión). Los procesos de activación plaquetaria comprenden complejas rutas de señalización que involucran sistemas de proteínas G acoplados a enzimas efectoras como la fosfolipasa A2, fosfolipasa C, adenilato y guanilato ciclasas. Las propiedades funcionales y estructurales de las plaquetas están estrechamente relacionadas, lo que les confiere un alto potencial trombogénico, contribuyendo sustancialmente en el desarrollo de un infarto cerebral. Coagulación 1: son un grupo de afecciones en las cuales hay un problema con el proceso de coagulación sanguínea del cuerpo. esto puede llevar a que se presente un sangrado intenso y prolongado.
PROBLEMAS DE COAGULACIÓN - 2 - Deficiencia del factor X Es un trastorno causado por la presencia de muy poca cantidad de una proteína llamada factor X en la sangre, lo cual lleva a problemas de coagulación.
Causas Cuando uno sangra, el cuerpo inicia una serie de actividades que ayudan a que la sangre se coagule, lo cual se denomina cascada de la coagulación. El proceso involucra proteínas especiales, llamadas factores de coagulación. Cuando falta uno o más de estos factores de coagulación, generalmente hay una posibilidad más alta de sangrado Trombo: es una masa intravascular o intracardíaca formada por elementos de la sangre e insolubles en ella, se caracterizan por estar fijos a la pared del vaso por una zona donde son más rígidos y consistentes. Pueden ser arteriales o venosos.
Coagulos: intravasculares son postmorten son gelatinosos, con una porción declive rojo oscura porque los eritrocitos sedimentan por acción de la graverdad y un sobrenadante amarillo en grasa de pollo, no estan fijos a la pared del vaso. En cambio los trombos son mas consistentes, estan anclados en un punto a la pared del vaso o cardiaca, los arteriales consistentes de color blanco grisaceo y friables, formados por una red de plaquetas fibrina y leucocitos en degeneración. Fibrinólisis. (De fibrina, y del griego lyein, disolver). La fibrinólisis primaria se refiere a la descomposición normal de los coágulos, mientras que la fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa. El sistema fibrinolítico va a jugar además un papel importante en diversas situaciones en las que se produce proteolisis tisular, tales como inflamación, invasión tumoral o neovascularización. La importancia fisiopatológica del sistema fibrinolítico deriva de que las alteraciones que condicionan un defecto de actividad fibrinolítica pueden predisponer a la trombosis, mientras que un exceso de activación favorecería la aparición de hemorragia.
Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro,1 derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos. Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva. Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebro vascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores. Cualquier anormalidad o enfermedad de las plaquetas se denomina trombocitopatía,2 la cual puede consistir, ya sea en tener un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Se pueden producir desórdenes que reducen el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos. Sin embargo, otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragias. Las plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular; Estos dos factores de crecimiento han demostrado jugar un papel significativo en la regeneración y reparación del tejido conectivo; Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.
coagulacion : Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre. Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional. El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse. Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas. Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo
La trombosis es un coágulo en el interior de un vaso sanguíneo y uno de los causantes de un infarto agudo de miocardio. También se denomina así al propio proceso patológico, en el cual, un agregado de plaquetas o fibrina ocluye un vaso sanguíneo. Cuando un vaso sanguíneo se lesiona, el cuerpo utiliza plaquetas (trombocitos) y fibrina para formar un coágulo de sangre para prevenir la pérdida de sangre. Incluso cuando un vaso sanguíneo no se lesione, los coágulos de sangre se pueden formar en el cuerpo en ciertas condiciones. Un coágulo que se desprende y comienza a viajar por todo el cuerpo se conoce como embolia.1 2 El trombo es una masa que se forma en el interior del aparato circulatorio y está constituida por la sangre del paciente, específicamente por los elementos sólidos de la sangre. En un individuo vivo la sangre puede coagularse pero esto es fuera del aparato circulatorio por ejemplo, la sangre que pasa al peritoneo, a la pleura o al pericardio. Ahí la sangre coagula y no es un trombo, porque está fuera del aparato cardiovascular. Las causas son: alteración en los vasos sanguíneos; arteriosclerosis ruptura traumática alteración en los factores de la coagulación; trombina, protrombina disminución de la Proteína C, Proteína S, llamadas estas últimas trombofilias. Los mecanismos que favorecen la formación de un trombo, son las alteraciones del flujo sanguíneo y estas alteraciones pueden deberse a reposo excesivo en cama (pacientes postoperados). Además en la intervención quirúrgica ha habido una estimulación de los factores de coagulación por la rotura de vasos, la sutura, una serie de intervenciones que involucran al aparato vascular. No es raro que un individuo se opere de una hernia inguinal, y en el momento que se le da de alta y empieza a moverse más de lo que se ha movido en los días anteriores presente una embolia fulminante ocasionándole la muerte. La tercera causa que influye son los componentes de la sangre. Cuando la sangre es más densa disminuyen los líquidos y aumentan los elementos figurados. O hay una hemoconcentración o una policitemia real. Dentro de este se incluye las trombosis a repetición. Los sitios de formación de trombo son en el corazón, arterias, venas y capilares, por lo que la trombosis puede formarse en cualquier parte del aparato circulatorio. Otras patologías que pueden provocar una trombosis son aquellas que presentan flujos en torbellinos, como las estrecheces valvulares. Un ejemplo es la estenosis mitral, en donde el flujo en la aurícula se hace más lento y favorece la trombosis. En la estenosis mitral hay que tener en cuenta que lo más probable es que haya trombosis en la orejuela y en alguna parte de la pared de la aurícula. Y si ésa hace flutter o fibrilación, la contracción de la aurícula es ineficiente. Entonces la aurícula no se contrae y además existe oclusión en la salida, de tal modo que hay una lentitud del flujo de salida y por tanto formación de coágulos (trombos). Otra causa de trombosis es el daño del endotelio. Si un vaso se inflama, por ejemplo una vena de las EEII por un trauma, se produce una lesión de la vecindad y daño endotelial, que desencadena inmediatamente la cascada de coagulación depositándose trombos en la superficie del vaso. En cuanto a los líquidos extraños que entran al aparato circulatorio y pueden provocar una embolia, estos pueden ser fundamentalmente el líquido amniótico que ingresa a la circulación materna al producirse desprendimiento de la placenta, y rotura de las venas uterinas y/o del cervix constituyendo en una embolia amniótica, ésta dependiendo de la cuantía puede ser fatal. También en las fracturas múltiples, la medula ósea adiposa de los huesos que es semilíquida puede entrar a la circulación y embolizar hacia el pulmón o cerebro.
La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP. Las células endoteliales producen una proteína llamada factor de von Willebrand,un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el factor de von Willebrand es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas. Cuando la capa endotelial es lesionada, el colágeno, el factor de von Willebrand y el factor tisular del endotelio son expuestos al flujo sanguíneo. Cuando las plaquetas hacen contacto con el colágeno o el factor de von Willebrand, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio. La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria(plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
Coagulación
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
El sistema plasmático de la coagulación, que está compuesto por una serie de factores: • Factor I o fibrinógeno • Factor II o protrombina • Factor III o tromboplastina tisular • Factor IV, que es el Ca++ • Factor V o proacelerina • Factor VII o proconvertina • Factor VIII o antihemofílico A • Factor IX o Christmas o factor antihemofílico B • Factor X o de Stuart- Prower • Factor XI o antecedente de la tromboplastina del plasma Factor XII o de Hageman • Factor XIII o estabilizador de la fibrina • Activador de la protrombina o tromboplastina completa Factor de Fitzgerald o kininógeno de alto peso molecular
Fibrinolisis
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro,1 derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos.
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre. Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Las plaquetas son producidas en el proceso de formación de las células sanguíneas llamado (trombopoyesis) en la médula ósea, por fragmentación en los bordes citoplasmáticos de los megacariocitos. El rango fisiológico de las plaquetas es de 150-400 x 109/litro. Alrededor de 1 x 1011 plaquetas de media son producidas cada día por un adulto sano. La expectativa de vida de las plaquetas circulantes es de 7 a 10 días. La producción de megacariocitos y plaquetas está regulada por la trombopoyetina, una hormona producida usualmente por el hígado y los riñones. Cada megacariocito produce entre 5.000 y 10.000 plaquetas. Las plaquetas son destruidas por fagocitosis en el bazo y por las células de Kupffer en el hígado. Una reserva de plaquetas es almacenada en el bazo y son liberadas cuando se necesitan por medio de contracción esplénica mediada por el sistema nervioso simpático. Formación de trombos
La función plaquetaria consiste en el mantenimiento de la hemostasia; Esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio.
Activación
La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP. Las células endoteliles producen una proteína llamada factor de von Willebrand (FvW),un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas. Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo. Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio. La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria; estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
Un enorme número de sustancias puede actuar sobre las plaquetas e inducir su activación [el factor de activación plaquetaria,6 trombina, el tromboxano (TXA2), fosfato de adenosina (ADP),10 epinefrina, vasopresina y serotonina] a través de sus respectivos receptores, pero todas lo hacen a merced de rutas de señalización intracelular que involucran un sistema de proteínas G acoplado a enzimas efectoras comolas fofolipasas A2 (PLA2) y C (PLC) y adenilato y guanilato ciclasas.5, 6, 10 Hasta ahora se han identificado nueve componentes diferentes de la familia de proteínas G en las plaquetas y, dependiendo del tipo de proteína G que participe en la señalización. Se puede activar o inhibir el efecto.6 Un ejemplo lo constituye la regulación por proteína G de la adenilato ciclasa y por tanto de la producción de monofosfato de adenosina cíclico (AMPc). Un incremento de las cantidades de AMPc bloquea la liberación de calcio en el STD e inhibe la función de la plaqueta. Este es el mecanismo que utilizan las prostaciclinas del endotelio vascular sano para inhibir la activación plaquetaria.
COAGULACION DE LA SANGRE: Es el proceso por el cual se forma un coágulo sanguíneo. Comienza en respuesta a una lesión en un vaso sanguíneo.
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso, por el cual, la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoléculares en forma de una red tridimensional.
El fibrinógeno, una vez transformado, recibe el nombre de fibrina. Coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse.
Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas.
Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc.
En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares.
A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos. Siete de los factores de coagulación (precalicreína —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para su síntesis en el hígado, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
Los coágulos sanguíneos (coágulos de fibrina) son las masas que se forman cuando la sangre se coagula. Los coágulos sanguíneos o de fibrina son masas que se forman cuando la sangre se coagula y un trombo es un coágulo de sangre que se forma en un vaso o dentro del corazón y permanece allí. Un émbolo es un trombo que viaja desde el vaso o la cámara del corazón donde se formó a otro lugar del cuerpo y el trastorno causado se llama embolia o embolismo (por ejemplo, embolia pulmonar).
Los trombos y émbolos pueden adherirse fírmemente a un vaso sanguíneo y bloquear parcial o completamente el flujo de sangre en dicho vaso. Esta obstrucción priva a los tejidos en esa área del flujo normal de sangre y oxígeno. Esta condición se llama isquemia y, de no tratarse rápidamente, puede ocasionar daño e incluso la muerte de los tejidos (infarto y necrosis) en dicha área.
La fibrinólisis primaria se refiere a la descomposición normal de los coágulos, mientras que la fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa.
Cuando se produce demasiado rápidamente, puede provocar hemorragias espectaculares (fibrinólisis hemorrágica) intra o postoperatorias (particularmente en Cirugía Torácica), o después de un parto, de un aborto o de un choque traumático; o bien puede provocar hemorragias menos importantes (hematuria) en el curso de ciertos cánceres (próstata, páncreas, estómago), de las cirrosis y de las leucosis. Obedece a la liberación de activadores de la profibrinolisina, o bien a la producción de un fermento proteolítico por los tejidos enfermos.
Puede ser primitiva aunque por lo general es secundaria, reactiva, en el curso de un síndrome de coagulación intravascular diseminada.
SISTEMA FIBRINOLITICO: El sistema fibrinolítico endógeno se activa por la presencia de un trombo intravascular, convirtiendo el plasminógeno en plasmina, sustancia con gran poder fibrinolítico. Esta reacción cuando la desencadenamos farmacológicamente puede ocurrir en todo el territorio circulatorio produciendo un estado de "lisis sistémica", o bien, ocurrir selectivamente a nivel del trombo de fibrina, dependiendo del fármaco empleado. Ninguno de estos fármacos diferencia entre un trombo oclusivo y un coágulo hemostásico, por lo que puede producir hemorragias a cualquier nivel si disuelve uno de estos coágulos.
La sangre circula por el interior de los vasos, los glóbulos rojos la más abundante masa celular sanguínea) lo hace por el centro del vaso, mientras que las plaquetas circulan más próximas a la superficie endotelial para vigilar la integridad del endotelio. El endotelio normal proporciona una superficie no adhesiva para las plaquetas. Cuando se rompe el endotelio, el colágeno expuesto favorece la adhesión de las plaquetas por medio de la interacción con una cantidad de receptores,pero es esencial para concretar la adhesión el Factor de Von Willebrand. A partir de esto y con el aumento del Ca++ intracelular se produce la activación de las plaquetas, lo que genera una serie de eventos que favorece la agregación plaquetaria, como son: su cambio de forma y de ser una esfera regular pasa a tener prolongaciones, liberación del contenido de los gránulo como el ADP que torna a las plaquetas pegajosas, el TX A2 derivado del Ácido Araquidónico que es vasoconstrictor y agregante plaquetario. El Factor II (trombina)
2,3,4,5 Coagulacion de la sangre 1,2,3,4
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc. A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina factor y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos. Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada. La cascada de coagulación se divide en tres vias: La via intrínseca, la vía extrínseca y la vía común.Las vías intrínseca y extrínseca son las vías de iniciación de la cascada, mientras que la vía común es hacia donde confluyen las otras dos desembocando en la conversión de fibrinógeno en fibrina lo cual sella y forma los trombos y coagulos.
6-Trombo
Cuando un vaso sanguíneo se lesiona, el cuerpo utiliza plaquetas y fibrina para formar un coágulo de sangre para prevenir la pérdida de sangre. Incluso cuando un vaso sanguíneo no se lesione, los coágulos de sangre se pueden formar en el cuerpo en ciertas condiciones. El trombo es una masa que se forma en el interior del aparato circulatorio y está constituida por la sangre del paciente, específicamente por los elementos sólidos de la sangre. Las causas son alteración en los vasos sanguíneos como arteriosclerosis,ruptura traumática y alteración en los factores de la coagulación
Es un proceso corporal normal que impide que los coágulos sanguíneos que ocurren en forma natural crezcan y causen problemas.La fibrinólisis primaria se refiere a la descomposición normal de los coágulos.La fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa. Esto puede provocar sangrado intenso.
8-Sistema fibrinolitico
La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina(PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón.La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca:En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias,segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA.En la vía intrínseca es la calicreína la encargada de mediar la activación del plasminógeno. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y elinhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
MECANISMOS DE AGREGACION Y ADHERENCIA PLAQUETARIA Las células endoteliales lesionadas liberan factor de von Willebrand y tromboplastina hística y endotelina (vasoconstrictor potente que reduce la pérdida de sangre). Las plaquetas se adhieren a la colágena subendotelial, liberan el contenido de sus gránulos y se adhieren unas a otras. Estos factores en conjunto se conocen como activación plaquetaria. La liberación de parte de sus contenidos granulares (en especial difosfato de adenosina (ADP) y trombospondina), torna a las plaquetas “pegajosas” y da lugar a que se adhieran las plaquetas circulantes a las plaquetas unidas a la colágena y se desgranulen. El ácido araquidónico, formado en el plasmalema de plaquetas activadas se convierte en tromboxano A2, un vasoconstrictor y activador de plaquetas potente. Las plaquetas agregadas actúa como un tapón que bloquea la hemorragia. Además expresan factor 3 plaquetario en su plasmalema, necesario para el ensamble apropiado de factores de la coagulación ( en especial de trombina). Tanto la tromboplastina hística como la tromboplastina plaquetaria actúa convirtiendo la protombina en trombina, la cual facilita la agregación plaquetaria. En presencia de calcio también convierte el fibrinógeno en fibrina. Los monómeros de fibrina que se producen forma un retículo de coágulo que conjunta plaquetas adicionales, eritrocitos y leucocitos en un coágulo sanguíneo (trombo). Alrededor de una hora después de formarse el coágulo los monómeros de actina y miosina forman filamentos delgados y gruesos , con lo cual se contrae el coágulo casi la mitad de su tamaño previo, tracciona los bordes del vaso, los acerca entre sí y minimiza la pérdida de sangre. Finalmente cuando se repara el vaso, las células endoteliales liberan activadores del plasminógeno que convierten el plasminógeno circulante en plasmina, la cual inicia la lisis del trombo.
MECANISMOS DE AGREGACION Y ADHERENCIA PLAQUETARIA Las células endoteliales lesionadas liberan factor de von Willebrand y tromboplastina hística y endotelina (vasoconstrictor potente que reduce la pérdida de sangre). Las plaquetas se adhieren a la colágena subendotelial, liberan el contenido de sus gránulos y se adhieren unas a otras. Estos factores en conjunto se conocen como activación plaquetaria. La liberación de parte de sus contenidos granulares (en especial difosfato de adenosina (ADP) y trombospondina), torna a las plaquetas “pegajosas” y da lugar a que se adhieran las plaquetas circulantes a las plaquetas unidas a la colágena y se desgranulen. El ácido araquidónico, formado en el plasmalema de plaquetas activadas se convierte en tromboxano A2, un vasoconstrictor y activador de plaquetas potente. Las plaquetas agregadas actúa como un tapón que bloquea la hemorragia. Además expresan factor 3 plaquetario en su plasmalema, necesario para el ensamble apropiado de factores de la coagulación ( en especial de trombina). Tanto la tromboplastina hística como la tromboplastina plaquetaria actúa convirtiendo la protombina en trombina, la cual facilita la agregación plaquetaria. En presencia de calcio también convierte el fibrinógeno en fibrina. Los monómeros de fibrina que se producen forma un retículo de coágulo que conjunta plaquetas adicionales, eritrocitos y leucocitos en un coágulo sanguíneo (trombo). Alrededor de una hora después de formarse el coágulo los monómeros de actina y miosina forman filamentos delgados y gruesos , con lo cual se contrae el coágulo casi la mitad de su tamaño previo, tracciona los bordes del vaso, los acerca entre sí y minimiza la pérdida de sangre. Finalmente cuando se repara el vaso, las células endoteliales liberan activadores del plasminógeno que convierten el plasminógeno circulante en plasmina, la cual inicia la lisis del trombo.
FORMACIÓN DEL TROMBO BLANCO Una vez lesionado el vaso, las plaquetas se adhieren al sitio dañado, que suele ser la superficie de la íntima al descubierto. La adherencia plaquetaria es mediada fundamentalmente por el factor de von Willebrand (von Willebrand factor, vWF), una gran proteína multimérica que está en el plasma y en la matriz extracelular de la pared subendotelial de los vasos, que actúa como el "adhesivo molecular primario", y genera potencia suficiente para soportar las grandes fuerzas de cizallamiento que actuarían para desprenderla con la corriente de sangre. La adherencia mencionada también es facilitada por la unión directa al colágeno subendotelial por los receptores específicos de dicha sustancia en la membrana plaquetaria. La adherencia plaquetaria origina activación y agregación de los trombocitos; dicho proceso es intensificado y amplificado por mediadores humorales en el plasma (como la adrenalina y la trombina); mediadores liberados de las plaquetas activadas (como el difosfato de adenosina y la serotonina) y los constituyentes de la matriz extracelular de la pared del vaso que se ponen en contacto con las plaquetas adherentes (como colágeno o vWF). Las plaquetas activadas pasan por una reacción de liberación en la cual secretan su contenido, que estimula aún más la agregación e inhibe los factores naturales anticoagulantes de las células del endotelio. En la agregación (interacción entre una plaqueta y otra) hay reclutamiento de más plaquetas desde la circulación hasta el sitio de la lesión vascular, con la cual se forma el llamado trombo blanco oclusivo, hecho de plaquetas, fijado y estabilizado con la trama de fibrina en evolución. El complejo de glucoproteína plaquetaria (Gp) IIb/IIIa (IIb3) es el receptor más abundante en la superficie de la plaqueta. La activación de los trombocitos transforma al receptor GpIIb/IIIa normalmente inactivo, en otra estructura activa que permite la unión al fibrinógeno y a vWF. La superficie de la plaqueta tiene aproximadamente 50 000 sitios de unión de fibrinógeno GpIIb/IIIa y por ello las innumerables plaquetas activadas reclutadas en el sitio de la lesión vascular rápidamente forman un "agregado" oclusivo por medio de una red densa de puentes de fibrinógeno intercelulares. El receptor en cuestión es el mediador básico de la agregación plaquetaria y por ello se ha vuelto un "elemento" eficaz para ser modificado por medio de terapia antiplaquetaria.
FORMACIÓN DEL COÁGULO DE FIBRINA Las proteínas de coagulación (factores de coagulación) circulan normalmente inactivas en el plasma. La serie de reacciones de coagulación que culminan en la formación de fibrina fue descrita originalmente como "cascada". Ya se describieron dos vías de coagulación de la sangre hace tiempo: la llamada vía extrínseca o de factor hístico, y la intrínseca o de activación por contacto. Se sabe ahora que la coagulación es iniciada normalmente al exponerse y activarse el factor hístico (tissue factor, TF), siguiendo la vía extrínseca clásica, pero interviene la amplificación importantísima por la participación de elementos de la vía intrínseca clásica. Dichas reacciones acaecen en las superficies fosfolípidas, por lo común en el exterior de la plaqueta activada. Los métodos para evaluar la coagulación en el laboratorio reflejan otros factores de influencia, por la naturaleza artificial de los sistemas in vitro usados. El elemento desencadenante inmediato de la coagulación es la lesión vascular, que expone la sangre al factor hístico (TF) que es expresado en forma constitutiva en las superficies de los componentes celulares del subendotelio de la pared, como las células de fibra lisa y los fibroblastos. El TF también aparece en micropartículas circulantes quizá desprendidas de células como los monocitos y las plaquetas. Dicho factor se liga al factor VIIa de proteasa serínica; el complejo activa el factor X y se transforma en Xa. Como otra posibilidad, el complejo indirectamente activa el factor X e inicialmente transforma el factor IX en IXa, y como paso siguiente activa el factor X. La participación del factor XI en la hemostasia no depende de su activación por el factor XIIa sino más bien de su activación retroalimentaria positiva por parte de la trombina. De este modo, el factor XIa actúa en la propagación y amplificación y no en el desencadenamiento de la cascada de la coagulación. El factor Xa, que se forma por las acciones del complejo de factor hístico/factor VIIa o el factor IXa (con el factor VIIIa como cofactor), transforma la protrombina en trombina, que es la proteasa fundamental del sistema de coagulación. El cofactor esencial de dicha reacción es Va. A semejanza de su homólogo el factor VIIIa, el factor Va es producido por la proteólisis limitada del factor V, inducida por trombina. La trombina es una enzima multifuncional que transforma el fibrinógeno plasmático soluble en una matriz insoluble de fibrina. La polimerización de la fibrina entraña un fenómeno ordenado de vínculos intermoleculares. La trombina también activa el factor XIII (factor estabilizante de fibrina) hasta la forma de factor XIIIa, que se enlaza en forma covalente y con ello estabiliza el coágulo de fibrina. El "ensamblado" de los factores de coagulación en las superficies activadas de la membrana celular acelera enormemente las reacciones que se suceden, y localiza la coagulación en sitios de lesión vascular. Los fosfolípidos ácidos, componentes fundamentales de la membrana celular, no están expuestos normalmente a las superficies de la membrana de la célula en reposo. Sin embargo, cuando la lesión vascular o estímulos inflamatorios activan plaquetas, monocitos y células endoteliales, los grupos "superiores" procoagulantes de los fosfolípidos aniónicos de la membrana son transpuestos a las superficies de tales células o liberados como parte de micropartículas, y quedan disponibles para apoyar y estimular las reacciones de coagulación plasmáticas.
EL SISTEMA FIBRINOLÍTICO La trombina que "escapa" a los efectos inhibidores de los sistemas anticoagulantes fisiológicos queda disponible para transformar el fibrinógeno en fibrina. En reacción a tal situación el sistema fibrinolítico endógeno es activado para "utilizar" la fibrina intravascular y con ello conservar o restablecer el libre tránsito de la circulación. De la misma forma que la trombina es la proteasa fundamental del sistema de coagulación, la plasmina lo es en el sistema fibrinolítico, al digerir la fibrina y generar sus productos de degradación. Los activadores de plasminógeno, que son el activador de tipo hístico (tissue type plasminogen activator, tPA) y el de tipo urocinasa (urokinase type plasminogen activator, uPA), separan Arg560-Val561 unida al plasminógeno, para generar la plasmina, enzima activa. Los sitios de unión lisínicos presentes en la plasmina (y el plasminógeno) permiten que se liguen a la fibrina, de tal manera que la fibrinólisis fisiológica es "fibrinoespecífica". El plasminógeno (por medio de sus sitios de unión lisínicos) y tPA poseen afinidad específica por la fibrina, y por lo tanto se ligan selectivamente a los coágulos. El ensamblado de un complejo ternario que consiste en fibrina, plasminógeno y tPA estimula la interacción localizada entre el plasminógeno y tPA, y acelera enormemente la activación de plasminógeno y su transformación en plasmina. Además, la degradación parcial de la fibrina por parte de la plasmina deja al descubierto nuevos sitios de plasminógeno y de unión con tPA en los residuos lisínicos de la terminación carboxilo de los fragmentos de fibrina, para intensificar todavía más tales reacciones. De esa manera, el organismo cuenta con un mecanismo extraordinariamente eficiente para generar en forma focal plasmina en el coágulo de fibrina, y de ese modo se transforma en sustrato de plasmina para la digestión, hasta la aparición de productos de degradación de la fibrina. La plasmina degrada la fibrina en sitios precisos de su molécula, de modo que se generan los característicos fragmentos de fibrina en el proceso de fibrinólisis. Los sitios de degradación son iguales que los correspondientes al fibrinógeno. Sin embargo, cuando la plasmina actúa en la fibrina con uniones covalentes, quedan en libertad los D-dímeros; en consecuencia, es posible medirlos en el plasma como un "índice" o reflejo relativamente específico de la degradación de fibrina (en vez de medirlos con fibrinógeno). Las mediciones de los D-dímeros se pueden utilizar como marcadores sensibles de la formación de coágulos, y algunos han sido validados para empleo en clínica, para así descartar el diagnóstico de trombosis venosa profunda (deep venous thrombosis, DVT) y embolia pulmonar en poblaciones escogidas. La regulación fisiológica de la fibrinólisis acaece más bien en dos niveles: 1) inhibidores del activador de plasminógeno (plasminogen activator inhibitors, PAI) en particular PAI1 y PAI2, con inhibición de los activadores de plasminógeno fisiológicos y 2) la antiplasmina 2 inhibe la plasmina. El PAI1 es el inhibidor primario de tPA y uPA en el plasma. La antiplasmina 2 es el principal inhibidor de la plasmina en el plasma de humanos e inactiva cualquier plasmina no vinculada con el coágulo de fibrina.
VIA INTRINSECA Se inicia por exposición del F. XII a superficies cargadas negativamente (membrana basal pared vaso). El factor XIIa activa la precalicreína a calicreína, que es un potente activador del factor XII (retroalimentación positiva). El F. XIIa, en la misma superficie, activa al factor XI. Estas dos reacciones requieren un cofactor no enzimático: el quininógeno de alto peso molecular (HMWK) que circula unido al factor XI y a la precalicreína. Finalmente el factor XIa activa al factor IX. Todas estas reacciones no requieren iones calcio.
Complejo procoagulante “tenasa” El factor IXa puede activar al factor X, pero de manera muy lenta. Requiere la presencia de su cofactor, el factor VIIIa. La activación del factor VIII la lleva a cabo la trombina. Una vez formado el VIIIa se genera el complejo tenasa entre el factor VIIIa(cofactor), el IXa (enzima) y el X (sustrato), sobre la superficie fosfolipídica cargada negativamente. Los tres factores se asientan sobre dicha superficie y exponen sus centros activos permitiendo una óptima activación del factor X. Esta superficie la aportan tanto plaquetas activadas como el endotelio dañado ( lo que localiza el coágulo en el sitio de rotura y controla su extensión).
Complejo “protrombinasa” A continuación el factor Xa (enzima) es liberado y se une al factor Va (cofactor), también activado por la trombina, y al factor II o protrombina (sustrato) generándose el complejo protrombinasa, necesitándose también la presencia de iones calcio y fosfolípidos.
Complejo procoagulante “tenasa extrínseca” La vía extrínseca se inicia tras el contacto con el factor tisular (FT) que no está normalmente expuesto al torrente circulatorio y que se libera como resultado de un daño endotelial. Ésta es la vía más relevante desde un punto de vista fisiopatológico. El FT se une al factor VII activándolo. El complejo FT-VIIa es capaz de generar trazas de factor Xa. Con lo que el complejo iniciador de la vía extrínseca es el formado por el factor VIIa (enzima), FT (cofactor), factor X (sustrato), iones calcio y una superficie fosfolipídica
Activación de los factores IX y X por el complejo factor VIIa-FT Sin embargo este proceso es relativamente lento y el complejo factor VIIa-FT-X también es capaz de activar al factor IX (vía intrínseca) con el fin de acelerar la formación del factor Xa. Por tanto el factor Xa es generado por dos vías diferentes. La 1ª, iniciada por el complejo FTVIIa, es más lenta, y ocurre en las primeras etapas, permitiendo la generación de trazas de trombina, que activará al factor VIII, permitiendo la formación del complejo tenasa y acelerando la producción de trombina. Por eso los factores VIII y IX son necesarios para una eficaz coagulación y por lo que las personas con déficits de estos factores sangran (hemofilia. El factor Xa se libera del complejo tenasa y se une al complejo protrombinasa, generando al factor IIa o trombina
VÍA COMÚN: trombina Además, la trombina activa a los factores V y VIII, a las plaquetas y al factor XIII, que se encarga de estabilizar el coágulo de fibrina. Y, además activa al factor XI, mediante una serie de reacciones de retroalimentación positiva. Al mismo tiempo, la trombina inicia también una de las vías anticoagulantes más potentes, la vía de la proteína C, lo que reduce la propia formación de trombina (retroalimentación negativa).
VÍA COMÚN: fibrinógeno Molécula formada por 2 subunidades idénticas. Cada subunidad formada por 3 cadenas glicoproteicas: Aα, Bβ, γ. La trombina libera un fragmento de las cadenas Aα y Bβ generando el monómero de fibrina y los fibrinopéptidos A y B. Ello genera cambios estructurales en el fibrinógeno que inducen su polimerización, tanto lineal como lateral, formando una malla tridimensional de fibrina inestable, que estabiliza el factor XIIIa (circula unido al fibrinógeno y es estimulado por la liberación de FPA y FPB). En la red de fibrina quedan también atrapadas células sanguíneas que hacen más impermeable al coágulo.
Fibrinolisis La fibrinolisis es un mecanismo esencial para eliminar los coágulos de fibrina durante el proceso de cicatrización, así como remover los coágulos intravasculares para impedir la trombosis. El efector final del sistema es la plasmina, que degrada la fibrina en productos de degradación (PDF y dímero D). La plasmina es producida a partir de un precursor inactivo, el plasminógeno, por acción de dos activadores del plasminógeno: activador tisular (t-PA) y activador tipo urocinasa (u-PA). La regulación de los activadores tiene lugar por la acción de inhibidores (PAI), de los que el más relevante es el PAI-1, mientras que la plasmina circulante es rápidamente inhibida por la α2-antiplasmina, lo que evita una fibrinolisis sistémica. La fibrinolisis se inicia por el t-PA liberado desde el endotelio en respuesta a diversos estímulos (trombina, oclusión venosa, ejercicio físico, etc). Una vez liberado se une a la fibrina donde activa el plasminógeno a plasmina que degrada la fibrina del coágulo. La trombina puede activar otro inhibidor fibrinolítico, el TAFI, el cual elimina residuos de lisina de la fibrina, lo que impide la unión del plasminógeno y ulterior degradación del coágulo.
Activación plaquetaria Un enorme número de sustancias puede actuar sobre las plaquetas e inducir su activación [el factor de activación plaquetaria,6 trombina, el tromboxano (TXA2), fosfato de adenosina (ADP),10 epinefrina, vasopresina y serotonina] a través de sus respectivos receptores, pero todas lo hacen a merced de rutas de señalización intracelular que involucran un sistema de proteínas G acoplado a enzimas efectoras comolas fofolipasas A2 (PLA2) y C (PLC) y adenilato y guanilato ciclasas.5, 6, 10 Hasta ahora se han identificado nueve componentes diferentes de la familia de proteínas G en las plaquetas y, dependiendo del tipo de proteína G que participe en la señalización. Se puede activar o inhibir el efecto.6 Un ejemplo lo constituye la regulación por proteína G de la adenilato ciclasa y por tanto de la producción de monofosfato de adenosina cíclico (AMPc). Un incremento de las cantidades de AMPc bloquea la liberación de calcio en el STD e inhibe la función de la plaqueta. Este es el mecanismo que utilizan las prostaciclinas del endotelio vascular sano para inhibir la activación plaquetaria. Los procesos de activación plaquetaria comprenden complejas rutas de señalización que involucran sistemas de proteínas G acoplados a enzimas efectoras como la fosfolipasa A2, fosfolipasa C, adenilato y guanilato ciclasas. Las propiedades funcionales y estructurales de las plaquetas están estrechamente relacionadas, lo que les confiere un alto potencial trombogénico, contribuyendo sustancialmente en el desarrollo de un infarto cerebral. Se realizó una revisión de las bases de la agregación, activación, degranulación plaquetaria y su participación en la enfermedad cerebrovascular. Se realizó una actualización del uso de los agentes antiplaquetarios y de la literatura actual referente al uso de bloqueadores de las glicoproteínas adhesivas de membrana
Son muchos los mediadores químicos relacionados con procesos inmunoinflamatorios y que desempeñan un papel importante tanto en afecciones locales como sistémicas.
De manera práctica podemos dividirlos en:
•Sustancias vasoactivas: bradiquinina, histamina, serotonina, etc. (Son mediadores preformados). •Derivados de fosfolípidos de membrana: prostaglandinas, leucotrienos y factor de activación plaquetaria (FAP). Estos son mediadores neoformados. •Productos de origen lisosomal: proteasas y proteínas catiónicas implicadas en la destrucción celular. • Radicales libres: anión superóxido y otros relacionados con la destrucción cellular.
Como toda molécula presente en los seres vivos, los radicales libres ejercen efectos útiles y efectos nocivos. Su papel de defensa durante la fagocitosis es positivo, la amplificación de estos fenómenos en la inflamación, trombosis o isquemia resulta perjudicial para las propias células y tejidos involucrados.
El factor de activación plaquetaria (FAP) es un potente mediador químico de la inflamación, es un derivado fosfolipídico involucrado en múltiples procesos patológicos relacionados con agregación plaquetaria, reacciones inmunoinflamatorias, trastornos vasculares y otros.
La biosíntesis del FAP se produce a partir de los ésteres de glicerilo de las membranas (glicerofosfolípidos). Mediante la fosfolipasa A2 (PLA2) se elimina el residuo acilo y se esterifica el alcohol en 2 (a menudo se trata de un ácido araquidónico), y queda conformado el liso-FAP. Una enzima del grupo de las transacetilasas, que utiliza la acetil coenzima A (acetil CoA), fija el acetilo en 2, y queda conformado el FAP que resulta muy activo y de vida extremadamente corta.
El liso-FAP es a la vez un precursor y un producto de degradación del FAP. No tiene éster acético fijado al alcohol 2 del glicerol y por lo tanto es inactivo. Parece que el liso -FAP puede circular de una célula a otra; por ejemplo, ir de las plaquetas a los neutrófilos y servir de precursor en la biosíntesis de FAP en estas células.
Coagulacion 1 El proceso hemostático comprende una fase relacionada con el vaso sanguíneo, otra con las plaquetas y otra en la que se produce la coagulación; se comprende que, para coagular la sangre, se precisa, ante todo, que la corriente sanguínea disminuya su propia velocidad y que la porción de pared de vaso lesionada quede como aislada, que se realice un cierre provisional (fase de las plaquetás) sobre la lesión, y que, por último, se establezca el definitivo (coagulación).
La capa de células que tapiza la superficie interna de los vasos se llama endotelio. En condiciones normales, se distribuyen por él cargas eléctricas negativas y, como también la superficie de las plaquetas está cargada negativamente, son rechazadas por la superficie interna de los vasos, repeliéndose entre sí. Cuando se lesiona un pequeño vaso (si son afectados los grandes vasos la teoría ya no tiene valor, puesto que la sangre sale con fuerza, debido a la elevada presión que existe en ellos, y entonces hay que detener la hemorragia artificialmente), como en el caso de una herida trivial, se produce una modificación de la carga eléctrica del endotelio en el punto lesionado. Entonces, las plaquetas se acumulan en gran número en este punto y se adhieren a la porción de superficie lesionada (donde se ha modificado el potencial eléctrico), formando un tapón, que cierra el punto abierto por el que escapa la sangre. Simultáneamente, la presión en el organismo desciende y disminuye también la fuerza con que la sangre escapa al exterior. Una vez que las plaquetas han entrado en contacto con la zona lesionada, empiezan a adelgazarse es decir, a disminuir de espesor, y a extenderse aumentando su diámetro. Paralelamente a este fenómeno, las plaquetas comienzan a desintegrarse. Mediante su destrucción se liberan diversas sustancias, entre las cuales está la serotonina, que provoca rápidamente una contracción de los vasos lesionados y de los que los rodean próximamente: de este modo, la sangría se estanca. La serotonina es una sustancia producida por unas células especiales del intestino, denoinadas células enterocromafinas; pasa luego a la corriente sanguínea, donde es absorbida por las plaquetas y liberada por éstas cuando se desintegran. Hay también otra sustancia (factor plaquetario) que, en presencia del calcio contenido habitualmente en el plasma contribuye a iniciar el proceso de coagulación.
Pero el coágulo, después de haberse formado, es todavía blando y friable, hasta que se produce una retracción, que lo transforma en una masa más compacta.
La coagulación es, en cierto sentido, un mecanismo de defensa de enorme utilidad para el organismo. En condiciones normales, tiene la misión concreta de limitar, hasta detenerlas, las pérdidas de sangre debidas a eventuales lesiones de los vasos sanguíneos. Este dispositivo, muy delicado y complejo, está confiado a unos factores que se encuentran en el plasma, y a las plaquetas, unos componentes celulares que figuran entre los "elementos corpusculares" de la sangre. En realidad, las plaquetas no son células, sino, simplemente, fragmentos de citoplasma (sin núcleo) de dos o tres milésimas de milímetro de diámetro, que se desprenden de células muy grandes, llamadas megacariocitos. Los megacariocitos residen en la médula ósea y, habitualmente, no penetran en la circulación; sólo los trocitos de citoplasma que se desprenden de ellos son transportados por la sangre. Las plaquetas sobreviven tres días o poco más, por término medio, y luego, si entre tanto no han sido utilizadas en el proceso de detención de una eventual hemorragia, sufren el mismo destino de los demás elementos de la sangre, es decir, son destruidos por las células del "sistema reticuloendotelial".
Los factores del plasma que entran en el mecanismo de la coagulación están representados por el fibrinógeno, la protrombina, el calcio y otras numerosas sustancias. Pero la coagulación no sería suficiente por sí sola para detener la salida de sangre de la lesión de un vaso. Aunque, en realidad, es el mecanismo más importante y perfeccionado, forma parte de un conjunto de fenómenos, orientados todos a detener la hemorragia, que toma el nombre de hemostasia.
El proceso de coagulación más propiamente dicho, consiste en la transformación de una sustancia presente en el plasma, llamada fibrinógeno, en otra llamada fibrina. Esta reacción se produce por obra de la trombina, que se forma a partir de la protrombina, bajo la influencia de la tromboplastina. En condiciones normales, no existe trombina (pero está presente un precursor inactivo suyo, la protrombina, que es una proteína de origen hepático) ni tromboplastina, pues de lo contrario, la sangre se coagularía habitualmente en los vasos con graves consecuencias.
Por lo tanto, en la coagulación podemos distinguir tres momentos: •1) formación de la tromboplastina • 2) transformación de la protrombina en trombina •3) transformación del fibrinógeno, proteína soluble en fibrina insoluble.
La formación de la tromboplastina es muy compleja; se debe a la activación de diversos factores presentes en el plasma y en los tejidos; la activación de uno de ellos repercute sobre el inmediatamente siguiente no activado todavía, como en una reacción en cadena.
El punto de partida de este proceso es doble: es decir, el proceso es puesto en marcha tanto por factores contenidos en el plasma, como por otros presentes en los tejidos (factores plasmáticos y factores tisulares), que se liberan a raíz de la salida de la sangre. Una vez formada, la tromboplastina actúa rápidamente sobre la protrombina y la transforma en trombina; de este modo, termina también la segunda fase de la coagulación y comienza la tercera, en la que la trombina actúa sigue y lo transforma en fibrina, sustancia, por el contrario, insoluble en el plasma (parte líquida de la sangre). El fibrinógeno es una gruesa proteína en forma de agujas, que se modifica bajo la acción de la trombina. Muchos elementos de fibrinógeno así modificados se unen por los extremos, dando lugar a larguísimos filamentos muy delgados. Éstos se reúnen en haces de mayor espesor, que se agrupan entre sí, formando una densa red visible de fibrina, entre cuyas mallas se encuentran aprisionados glóbulos rojos, glóbulos blancos y restos de plaquetas. Este es el coágulo.
Existen dos vías llevan a la formación de un coágulo de fibrina: la vía intrínseca y la vía extrínseca. Aunque las dos son iniciadas por mecanismos diferentes, las dos convergen en una vía común que lleva a la formación del coágulo. La formación del trombo rojo ó coágulo en respuesta a una anormalidad en un vaso pero en la ausencia de una lesión del tejido es el resultado de la vía intrínseca.
La vía intrínseca tiene poca significancia bajo condiciones fisiológicas normales. El suceso más importante clínicamente es la activación de la vía intrínseca por el contacto de la pared del vaso con las partículas de lipoproteína, VLDLs y quilomicrones. Este proceso claramente demuestra el papel de la hiperlipidemia en la generación de la ateroesclerosis. La vía intrínseca también puede ser activada por el contacto de la pared del vaso con bacterias.
La formación del coágulo de fibrina en respuesta a una lesión del tejido es el evento clínico más importante de la hemostasis bajo condiciones normales de respuesta. Este proceso es el resultado de la activación de la vía extrínseca.
Ambas vías son complejas e incluyen varias proteínas diferentes denominadas factores de la coagulación.
Trombo y Coágulo Un trombo o coágulo de la sangre, es el producto final de la etapa de coagulación de la sangre en la hemostasis. Esto se logra a través de la agregación de las plaquetas que forman un tapón de plaquetas, y la activación del sistema de coagulación humoral (es decir, los factores de coagulación). Un trombo es normal en los casos de lesiones, pero patológica en casos de trombosis.
Son los trombos murales trombos adherente a la pared del vaso. Ellos no son oclusivos y afectan a los vasos grandes, tales como el corazón y la aorta. Macroscópicamente que aparecen en gris-rojo con líneas claras y oscuras (líneas de Zahn) que representan bandas de fibrina (más ligero) con las células blancas de la sangre atrapados y las células rojas de la sangre (más oscuro) alterna.
En concreto, un trombo es la activación inapropiada del proceso hemostático en un recipiente sin lesiones o heridas leves. Un trombo en un vaso sanguíneo grande disminuirán el flujo de sangre a través de ese recipiente (denominado un trombo mural). En un pequeño vaso sanguíneo, el flujo sanguíneo puede ser completamente de corte (denominado un trombo oclusivo) que resulta en la muerte de tejido suministrado por dicho buque. Si se desplaza un trombo y se convierte en libre flotación, que se denomina como un émbolo.
Algunas de las condiciones que elevan el riesgo de coágulos sanguíneos en desarrollo incluyen la fibrilación auricular (un tipo de arritmia cardiaca), reemplazo de válvula cardíaca, un reciente ataque al corazón (también llamado infarto de miocardio), períodos largos de inactividad, y deficiencias genéticas o enfermedades relacionadas en las capacidades de coagulación de la sangre.
87462 ESTEBAN M GUERRERO P DIAPOSITIVAS TODAS La habilidad del cuerpo para controlar el flujo de sangre luego de una lesión vascular es un componente indispensable de la supervivencia. El proceso de la coagulación sanguínea y luego la disolución del coágulo, seguido por una reparación del tejido lesionado, se denomina hemostasis. La hemostasis se conforma de 4 eventos principales que ocurren en un orden determinado luego de la pérdida de la integridad vascular:
1. La fase inicial del proceso es la constricción vascular. Esto limita el flujo sanguíneo al área de la lesión.
2. A continuación, se activan las plaquetas por la trombina y se agregan en el sitio de la lesión, formando un tampón temporario y flojo conformado de plaquetas. La proteína fibrinógeno es principalmente responsable de estimular la agregación plaquetaria. Las plaquetas se agregan al unirse al colágeno que se expone debido a la ruptura del recubrimiento epitelial de los vasos. Luego de su activación, las plaquetas liberan ADP un nucleótido y un eicosanoide, TXA2 (los cuales activan más plaquetas), serotonina, fosfolípidos, lipoproteínas y otras proteínas importantes de la cascada de coagulación. Además de su secreción, las plaquetas activadas cambian su conformación para acomodar la formación del coágulo.
3. Para asegurar la estabilidad del tampón flojo inicial, se forma una malla de fibrina (también llamada un coágulo) que recubre al tampón. Si el tampón únicamente contiene plaquetas se denomina un trombo blanco; si glóbulos rojos están presentes se lo denomina un trombo rojo
4. Finalmente, el coágulo debe ser disuelto para que el flujo sanguíneo normal pueda resumir luego de que se repare el tejido. La disolución del coágulo ocurre a través de la acción de la plasmina Dos vías llevan a la formación de un coágulo de fibrina: la vía intrínseca y la vía extrínseca. Aunque las dos son iniciadas por mecanismos diferentes, las dos convergen en una vía común que lleva a la formación del coágulo. La formación del trombo rojo ó coágulo en respuesta a una anormalidad en un vaso pero en la ausencia de una lesión del tejido es el resultado de la vía intrínseca. La vía intrínseca tiene poca significancia bajo condiciones fisiológicas normales. El suceso más importante clínicamente es la activación de la vía intrínseca por el contacto de la pared del vaso con las partículas de lipoproteína, VLDLs y quilomicrones. Este proceso claramente demuestra el papel de la hiperlipidemia en la generación de la ateroesclerosis. La vía intrínseca también puede ser activada por el contacto de la pared del vaso con bacterias.
La formación del coágulo de fibrina en respuesta a una lesión del tejido es el evento clínico más importante de la hemostasis bajo condiciones normales de respuesta. Este proceso es el resultado de la activación de la vía extrínseca.
Ambas vías son complejas e incluyen varias proteínas diferentes denominadas factores de la coagulación.
87462 ESTEBAN M GUERRERO P DIAPOSITIVAS TODAS Activación Plaquetaria y el Factor von Willebrand (vWF)
Para que ocurra la hemostasis, las plaquetas deben adherirse al colágeno expuesto, liberar los contenidos de sus gránulos y agregarse. La adhesión plaquetaria al colágeno expuesto en las superficies endoteliales de las células es mediada por el factor von Willebrand (vWF). Las deficiencias hereditarias del vWF son la causa de la enfermedad de von Willebrand, (vWD) (ver abajo para más detalles). La función del vWF is actuar como un puente entre un complejo específico de glicoproteínas en la superficie de las plaquetas (GPIb-GPIX-GPV) y las fibrillas de colágeno. El GPIb parte del complejo se compone de dos proteínas, GPIbα y GPIbβ codificadas por los genes por separado. La importancia de esta interacción entre el vWF y el complejo GPIb de las plaquetas es ilustrado en los desórdenes hereditarios de la coagulación causados por defectos en las proteínas del complejo GPIb, de los cuales el más común es el síndrome de Bernard-Soulier (también conocido como el síndrome de las plaquetas gigantes).
Además de servir como un puente entre las plaquetas y el colágeno expuesto de las superficies endoteliales, el vWF se une y estabiliza el factor de coagulación VIII. La unión del factor VIII por el vWF es necesaria para la supervivencia normal del factor VIII en la circulación.
El factor von Willebrand es una glicoproteína multimérica compleja que es producida y almacenada en los gránulos α de las plaquetas. También es sintetizado por los megacariocitos y se encuentra asociado al tejido conectivo subendotelial.
La activación inicial de las plaquetas es inducida por una trombina ligada a un receptor específico ubicado en la superficie de las plaquetas, lo cual inicia la transducción de una cascada de señalización. El receptor de la trombina está acoplado a una proteína G que a su vez activa a la fosfolipasa C-γ (PLC-γ). La PLC-γ hidroliza al fosfatidilinositol-4,5-bifosfato (PIP2) resultando en la formación de inositol trifosfato (IP3) y diacilglicerol (DAG). El IP3 induce la liberación de los almacenes intracelulares de Ca2+ y el DAG activa a la proteína cinasa C (PKC).
El colágeno al cual se adhieren las plaquetas al igual que la liberación del Ca2+ intracelular resulta en la activación de la fosfolipasa A2 (PLA2) la cual a continuación hidroliza fosfolípidos en la membrana, llevando a la liberación del ácido araquidónico. La liberación del ácido araquidónico resulta en un aumento en la producción y liberación del tromboxano A2 (TXA2). El TXA2 es un vasoconstrictor potente e induce la agregación plaquetaria y funciona al unirse a receptores que trabajan a través de la vía de la PLC-γ.
Otra enzima que es activada por la liberación del Ca2+ intracelular es la cinasa de la cadena liviana de miosina myosin light chain kinase (MLCK). La MLCK activada fosforila a la cadena liviana de la miosina la cual interactúa con la actina, resultando en una alteración en la morfología y motilidad plaquetaria.
Uno de los varios efectos de la PKC es la fosforilación y activación de una proteína plaquetaria específica de 47,000 Daltons. Esta proteína activada induce la liberación de los contenidos de los gránulos plaquetarios; uno de los cuales es el ADP. El ADP promueve la estimulación plaquetaria al incrementar la activación general de la cascada. El papel importante del ADP en la activación plaquetaria puede apreciarse con el uso de antagonistas del receptor del ADP como el Plavix® (clopidogrel), en el control de la trombosis (ver abajo).
87462 ESTEBAN M GUERRERO P DIAPOSITIVAS TODAS El ADP también modifica la membrana de las plaquetas conllevando a una exposición del complejo de receptores de glicoproteínas plaquetarias: GPIIb-GPIIIa. El GPIIb-GPIIIa constituye un receptor para el vWF y el fibrinógeno, resultando en una agregación plaquetaria inducida por el fibrinógeno. El complejo GPIIb-GPIIIa es un miembro de una familia de integrinas de receptores de la superficie celular que interactúan con la matríz extracelular. El complejo GPIIb-GPIIIa también es llamado αIIb-β3 integrina. La importancia del GPIIb-GPIIIa en la activación plaquetaria y la coagulación es ilustrada por el hecho de que los desórdenes de coagulación resultan de defectos heredados en este complejo de glicoproteínas. El más común de estas disfunciones plaquetarias hereditarias es la trombastenia de Glanzmann la cual resulta de defectos en la proteína GPIIb de este complejo. Además, la importancia de este complejo en la hemostasis es demostrada por el uso de anticuerpos que bloquean este receptor y actúan como anti-coagulantes (p. ej. ReoPro®, abciximab: ver abajo).
La activación de las plaquetas es necesaria para su consecuente agregación y formación del coágulo plaquetario. Sin embargo, igualmente de importante en la activación de la cascada de a coagulación es el papel de los fosfolípidos activados en la superficie de las plaquetas. Las cascadas de coagulación: la cascada intrínseca (la cual tiene menos importancia in vivo en comparación a la cascada extrínseca) es iniciada cuando se realiza el contacto entre la sangre y las superficies expuestas de carga negativa. La vía extrínseca es iniciada cuando ocurre una lesión vascular lo cual resulta en una exposición del factor tisular (TF) (también identificado como el factor III), una glicoproteína subendotelial en la superficie celular que se une a fosfolípidos. La flecha punteada verde representa un punto de cruce entre la vía extrínseca y la intrínseca. Las dos vías se convergen en la activación del factor X a Xa. El factor Xa tiene un papel en la activación del factor VII a VIIa como se demuestra por la flecha verde. El factor Xa activo hidroliza y activa la protrombina a trombina. La trombina puede por ende activar los factores XI, VIII y V lo cual ayuda a que proceda la cascada. Básicamente, el papel de la trombina es convertir el fibrinógeno a fibrina y activar el factor XIII a XIIIa. El factor XIIIa (también llamado transglutaminasa) se une a polímeros de fibrina y así solidificando el coágulo. HMWK = quininógeno de alto peso molecular. PK = precalicreina. PL = fosfolípidos. El Kallikrein-Kinin Sistema de Coagulación
El kallikrein-kinin sistema comprende un complejo de proteínas que, cuando activado conduce a la liberación de kinins vasoactivos. El kinins son liberados de alto peso molecular kininogen (HMWK) y de bajo peso molecular kininogen (LMWK) como consecuencia de la activación de cualquiera de los tejidos kallikrein o plasma kallikrein. El kallekreins sí existen en la pre-formas inactivas. El kinins están involucrados en muchos fisiológicos y patológicos incluidos los procesos de regulación de la presión arterial y el flujo (a través de la modulación de la renina-angiotensina vía), la coagulación de la sangre, la proliferación celular y el crecimiento, la angiogénesis, apoptosis, y la inflamación. Kinin acción en las células endoteliales conduce a la vasodilatación, el aumento de la permeabilidad vascular, la liberación de activador tisular del plasminógeno (tPA), la producción de óxido nítrico (NO), y la movilización de ácido araquidónico, fundamentalmente el resultado de la prostaciclina (PGI2) la producción por las células endoteliales. Aunque las actividades de la kallikrein-kinin sistema están involucrados en numerosos procesos, en esta sección se ocupará sólo con su función en la coagulación de la sangre. Con respecto a la hemostasia más importante es la bradiquinina kinin que se libera de HMWK.
La fibrinólisis es un proceso que previene los coágulos de sangre de crecimiento y convertirse en problemática [1] Este proceso tiene dos tipos:. Fibrinolisis primaria y fibrinólisis secundaria. El tipo principal es un proceso normal del cuerpo, mientras que la fibrinólisis secundaria es la descomposición de los coágulos debido a un medicamento, un trastorno médico, o alguna otra causa.
En la fibrinólisis, un coágulo de fibrina, el producto de la coagulación, se descompone. [2] Su plasmina principal enzima corta la malla de fibrina en varios lugares, que conduce a la producción de fragmentos circulantes que se eliminan por otras proteasas o por el riñón y el hígado .
La plasmina se produce en una forma inactiva, el plasminógeno, en el hígado. Aunque plasminógeno no puede escindir la fibrina, que todavía tiene una afinidad por ella, y se incorpora en el coágulo cuando se formó.
Activador del plasminógeno tisular (t-PA) [3] y uroquinasa son los agentes que convierten el plasminógeno en la plasmina activa, permitiendo de este modo que se produzca la fibrinólisis. t-PA se libera en la sangre muy lentamente por el endotelio dañado de los vasos sanguíneos, de tal manera que, después de varios días (cuando el sangrado se ha detenido), el coágulo se descompone. Esto ocurre porque plasminógeno se convirtió en atrapado dentro del coágulo cuando se formó, ya que se activa lentamente, se descompone la malla de fibrina. t-PA y uroquinasa se sí mismos inhibidas por el inhibidor del activador del plasminógeno-1 y el inhibidor del activador del plasminógeno-2 (PAI-1 y PAI-2). En contraste, la plasmina estimula aún más la generación de plasmina mediante la producción de formas más activas de tanto activador del plasminógeno tisular (tPA) y la uroquinasa.
Alfa 2-antiplasmina y alfa 2-macroglobulina inactivan plasmina. Actividad de la plasmina también se reduce por el inhibidor de la fibrinólisis activable por trombina (TAFI), que modifica la fibrina para que sea más resistente a la del plasminógeno mediada por tPA.
87462 ESTEBAN M GUERRERO P DIAPOSITIVAS TODAS Las dos formas de prekallikrein, plasma y tejidos, se derivan de diferentes los genes en diferentes cromosomas. El plasma kallikrein gen (símbolo KLKB1) está en el cromosoma 4q34–q35 y el tejido kallikrein gen (símbolo KLK1) situado en el cromosoma 19q13.2–q13.4. Estos dos son serina proteasas kallikreins cuyos principales sustratos son HMWK (plasma kallikrein) y LMWK (tejido kallikrein). Cuándo plasma prekallikrein está activada para kallikrein que Cleaves HMWK en dos fases proceso que, en definitiva, libera bradiquinina. Bradiquinina es un 9-aminoácido péptido vasoactivo que induce a la vasodilatación y el aumento vasculares permeabilidad. Activado tejido kallikrein Cleaves lysyl-bradiquinina (también llamado kallidin) de LMWK. Lysyl-bradiquinina es bradiquinina con un residuo de la lisina en la N-terminal que un 10-aminoácido péptido vasoactivo. Sus actividades son esencialmente idénticos a los de la bradiquinina.
Ambos HMWK y LMWK se derivan del mismo gen en el cromosoma 3q26–qter que está compuesto de 11 exones. Exones 1 al 9 de codificar la cadena pesada de ambas kininogens. Exón 10 codifica bradiquinina, así como la luz de la cadena de HMWK. Exón 11 codifica la cadena de luz LMWK. La pesada cadena de la luz y la nomenclatura se refiere a la servidumbre-disulfuro de la estructura de cada kininogen después de su a través de la activación por kallikrein escisión.
HMWK se considera una α-globulina y se compone de seis áreas funcionales. La proteína que circula en el plasma como una sola cadena polipeptídica con un peso molecular de 88–120 kDa depende el nivel de glucosilación. El cadena pesada es de 64 kDa y contiene los dominios 1, 2 y 3 mientras que la cadena de luz es de 45–56 kDa y comprende dominios 5 y 6. La cadenas pesadas y ligeras son unidos entre sí a través de dominio 4, que también contiene la secuencia de la bradiquinina. Dominio 1 contiene una baja afinidad de unión de calcio. Dominios 2 y 3 contienen secuencias de aminoácidos (QVVAG) que inhiben la cisteína proteasas. Dominio 3 también ha de plaquetas y células endoteliales vinculante actividad. Dominio de las secuencias de 5 ha heparina vinculante, sitios de unión celular, propiedades y antiangiogénicos. La unión de HMWK a las superficies de carga negativa se produce a través de una región de la histidina cadena de luz que se encuentra en dominio 5. 6 contiene el dominio prekallikrein y factor XI-sitios de unión. Al ser capaces de obligar a cargado a través de superficies de dominio 5 y al mismo tiempo obligar factor XI y prekallikrein dominio a través de 6, puede servir como HMWK cofactor en la activación de contacto con el plasma. LMWK se considera una β-globulina y tiene un peso molecular de 50 a 68 kDa. La estructura de LMWK es similar a la que de HMWK, sin embargo, la cadena de luz está a sólo 4–5 kDa y no tiene ningún contacto activación ni prekallikrein-sitios de unión.
El plasma kinin que forman el sistema se llama el sistema de contacto y de plasma se compone de factor XII, factor XI, prekallikrein y HMWK. Factor XII, prekallikrein, y HMWK saturably reversible y se unen a las células endoteliales, plaquetas y granulocitos en una reacción dependiente de zinc. Cuando se hace de plasma contacto con una superficie de carga negativa, el factor XII se une y se autoactivated del factor XII (el "uno" significa el factor activado). Entonces el factor XIIa prekallikrein activa a kallikrein y kallikrein Cleaves HMWK liberación bradiquinina. Existe también la activación del factor de reciprocidad XII por kallikrein resultante en el sistema de amplificación.
87462 ESTEBAN M GUERRERO P DIAPOSITIVAS TODAS Cascada Intrínseca de la Coagulación
La vía intrínseca (también llamada la vía de activación por contacto) es mucho menos significante en la hemostasis bajo condiciones fisiológicas normales en comparación a la vía extrínseca. Sin embargo, durante un estado patológico tal como la hiperlipidemia o una infiltración bacteriana puede conllevar a la activación de la trombosis a través de la cascada de coagulación de la vía intrínseca.
La vía intrínseca requiere de los factores de coagulación VIII, IX, X, XI y XII. También requiere de proteínas tales como la precalicreina (PK) y el quininógeno de alto peso molecular (HK o HMWK), al igual que iones de calcio y fosfolípidos secretados por las plaquetas. Cada uno de los componentes de estas vías resulta en la conversión del factor X (inactivo) al factor Xa ("a" significa activo). La iniciación de la vía intrínseca ocurre cuando la precalicreina, quininógeno de alto peso molecular, factor XI y el factor XII son expuestos a una superficie de carga negativa. A esto se lo denomina como la fase de contacto y puede ocurrir como el resultado de una interacción con los fosfolípidos (principalmente fosfotidiletanolamina, PE) de las partículas lipoproteínas circulantes tales como los quilomicrones, VLDL y LDL oxidada. Esta es la base del papel de la hiperlipidemia en promover un estado pro-trombótico y en el desarrollo de la arterosclerosis.
La activación de contacto de la vía intrínseca también puede ocurrir en la superficie de las bacterias, y mediante la interacción con el ácido úrico cristales, ácidos grasos, protoporfirina, β amiloide, y homocisteína. De hecho, los niveles elevados de homocisteína en la sangre han demostrado que son equivalentes con disfunción cardiovascular. Por lo tanto, es importante garantizar que la función apropiada de la reacción metionina sintasa se mantiene. Aunque sería suponer que una mayor ingesta de vitamina B12 debería conducir a Conversión de aumento de la homocisteína en metionina, y por lo tanto reduce los niveles de circulantes de homocisteína, estudios controlados han demostrado que esto no ocurrir.
El ensamblaje de los componentes de la fase de contacto resulta en la conversión de la precalicreina a la calicreina, la cual a su vez activa al factor XII a factor XIIa. El factor XIIa puede entonces hidrolizar más precalicreina a calicreina, estableciendo una recíproca activación de la cascada. El factor XIIa también activa al factor XI a factor XIa y conlleva a la liberación de bradicinina, un vasodilatador potente, a partir del quininógeno de alto peso molecular.
En la presencia del Ca2+, el factor XIa activa al factor IX a factor IXa. El factor IX es una proenzima que contiene residuos γ-carboxiglutamato (gla) vitamina K-dependientes, cuya actividad de proteasas de serina es activada luego de que el Ca2+ se une a los residuos gla. Varias de estas proteasas de serina de la cascada (II, VII, IX y X) son proenzimas que contienen gla. El factor activo IXa cliva al factor X en una unión interna de arg-ile (R-I) resultando en su propia activación al factor Xa.
La activación del factor Xa requiere el ensamblaje del complejo de tenasa (Ca2+ y los factores VIIIa, IXa y X) en la superficie de las plaquetas activadas. Una de las respuestas de las plaquetas a su activación es la presentación de un fosfatidilserina (PA) y un fosfatidilinositol (PI) en sus superficies. La exposición de estos fosfolípidos permite que se forme el complejo de tenasa. El papel del factor VIII en este proceso es de servir como un receptor, en la forma del factor VIIIa, para los factores IXa y X. El factor VIIIa se denomina un cofactor en la cascada de coagulación. La activación del factor VIII al factor VIIIa (el verdadero receptor) ocurre en la presencia de cantidades diminutas de trombina.
El sistema fibrinolítico sanguíneo comprende una proenzima inactiva, plasminógeno, que se puede convertir a la enzima activa, plasmina. La plasmina degrada la fibrina en productos de degradación de fibrina solubles, por los dos activadores del plasminógeno fisiológicas (PA), el tipo de tejido PA (t-PA) y el PA de tipo urocinasa (u-PA). mediada por la activación del plasminógeno de t-PA está implicado principalmente en la disolución de la fibrina en la circulación. u-PA se une a un receptor celular específico (u-PAR), resulta en una mayor activación de células del plasminógeno unido. La inhibición del sistema fibrinolítico puede ocurrir ya sea en el nivel de la PA, por los inhibidores de activador de plasminógeno específico (PAI), o en el nivel de la plasmina, principalmente por alfa-2 antiplasmina. Varias interacciones moleculares se han observado entre el sistema de las metaloproteinasas de matriz (MMP) y fibrinolítico, ambos sistemas pueden colaborar en la generación de actividad proteolítica. Por lo tanto, estromelisina-1 (MMP-3) escinde un fragmento de 55 kDa kringle 1-4, que contiene el sitio de unión de lisina (s) implicada en la unión celular, a partir de plasminógeno y elimina un 17-kDa fragmento NH2-terminal, que contiene el celular El sitio de unión al receptor, de uroquinasa (u-PA). De este modo, MMP-3 puede regular a la baja actividad de la plasmina asociada celular por la disminución de la cantidad de plasminógeno activable, sin afectar célula vinculada actividad de u-PA.
87462 ESTEBAN M GUERRERO P DIAPOSITIVAS TODAS Cascada Extrínseca de la Coagulación
El factor Xa activado es el sitio en el cual las cascadas de coagulación intrínseca y extrínseca se convergen. La vía extrínseca es iniciada en el sitio de la lesión en respuesta a la liberación del factor tisular (factor III) y por ende, es también conocida como la vía del factor tisular. El factor tisular es un cofactor en la activación catalizada del factor X por el factor VIIa. El factor VIIa una proteasa de serina que contiene un residuo gla, cliva al factor X en factor Xa de manera idéntica a la del factor IXa en la vía intrínseca. La activación del factor VII ocurre a través de la acción de la trombina o el factor Xa. La habilidad del factor Xa de activar al factor VII crea una asociación entre las vías intrínseca y extrínseca. Una asociación adicional entre las dos vías se da a través de la habilidad del factor tisular y el factor VIIa de activar al factor IX. Se cree que la formación del complejo entre el factor VIIa y el factor tisular es el paso principal en toda la cascada de coagulación. La evidencia de esta declaración nace del hecho que personas con deficiencias hereditarias en los componentes de la fase de contacto de la vía intrínseca no exhiben problemas de coagulación. Uno de los mecanismos más importantes de la inhibición de la vía extrínseca ocurre en el complejo tisular factor-factor VIIa–Ca2+–Xa. La proteína, un inhibidor de la coagulación asociado a una lipoproteína, LACI se une específicamente a este complejo. LACI también se denomina el inhibidor de la vía extrínseca, EPI o factor tisular inhibidor de la vía, TFPI y anteriormente se lo conocía como anticonvertina. LACI es compuesto de 3 dominios inhibidores de proteasas. El dominio 1 se une al factor Xa y el dominio 2 se une al factor VIIa pero sólo en la presencia del factor Xa.
Regreso al inicio Activación de la Protrombina a Trombina
El punto en el cual las dos vías se convergen es en la activación del factor X al factor Xa. El factor Xa activa a la protrombina (factor II) para convertirse en trombina (factor IIa). La trombina, a su vez, convierte el fibrinógeno a fibrina. La activación de la trombina ocurre en la superficie de las plaquetas activadas y requiere de la formación de un complejo de protrombinasa. Este complejo está compuesto de los fosfolípidos de las plaquetas: fosfatidilinositol y fosfatidilserina, Ca2+, factores Va y Xa y protrombina. El factor V es un cofactor en la formación del complejo de protrombinasa y tiene un papel similar al del factor VIII en la formación del complejo de tenasa. Como la activación del factor VIII, el factor V es activado al factor Va a través de cantidades diminutas y es inactivado por niveles aumentados de trombina. El factor Va se une a receptores específicos en la superficie de las plaquetas activadas y forma un complejo con la protrombina y el factor Xa.
La protrombina es una proteína de cadena simple de 72,000 Daltons, que contiene diez residuos gla en la región de su N-terminal. Dentro del complejo de protrombinasa, la protrombina es clivada en 2 sitios por el factor Xa. Este clivaje genera una molécula de trombina activa de 2 cadenas que contiene una cadena A y una cadena B las cuales están unidas por una unión simple de disulfuro.
Además de su papel en la activación de la formación del tampón de fibrina, la trombina juega un papel regulador importante en la coagulación. La trombina se une con la trombomodulina presente en la superficie de las células endoteliales formando un complejo que convierte la proteína C a la proteína Ca. La proteína S y la proteína Ca son cofactores que degradan a los factores Va y VIIIa y así limitando la actividad de estos 2 factores en la cascada de la coagulación.
87462 ESTEBAN M GUERRERO P DIAPOSITIVAS TODAS Activación del Fibrinógeno a Fibrina
El fibrinógeno (factor I) consiste en 3 pares de polipéptidos ([Aα][Bβ][γ])2. Las 6 cadenas están covalentemente unidas cerca de sus N-terminales a través de uniones disulfuro. Las porciones A y B de las cadenas Aα y Bβ constituyen los fibrinopéptidos, A y B, respectivamente. Las regiones de fibrinopéptidos del fibrinógeno contienen varios residuos de glutamato y aspartato impartiendo una alta carga negativa a esta región y promoviendo la solubilidad del fibrinógeno en el plasma. La trombina activa es una proteasa de serina que hidroliza al fibrinógeno en cuatro uniones arg-gly (R-G) entre el fibrinopéptido y las porciones a y b de la proteína.
La liberación de los fibrinopéptidos mediada por la trombina, genera monómeros de fibrina con una estructura de subunidades (αβγ)2. Estos monómeros se agregan espontáneamente en un arreglo regular, formando un coágulo de fibrina relativamente débil. Además de la activación de la fibrina, la trombina convierte el factor XIII al factor XIIIa, una transglutaminasa altamente específica que introduce uniones covalentes entre el nitrógeno de amidas de las glutaminas y el grupo ε-amino de las lisinas pertenecientes a los monómeros de fibrina.
Regreso al inicio Disolución de los Coágulos de Fibrina
La degradación de los coágulos de fibrina es la función de la plasmina, una proteasa de serina que circula como la proenzima inactiva plasminógeno. Cualquier plasmina libre circulante es rápidamente inhibida por la antiplasmina α2. El plasminógeno se une al fibrinógeno y a la fibrina, y así se incorporan en un coágulo. El activador tisular del plasminógeno (tPA) y, a un grado menor, la urocinasa son proteasas de serina que convierten el plasminógeno a plasmina. El tPA inactivo es liberado de las células endoteliales vasculares luego de una lesión; se une a la fibrina y es consecuentemente activado. La urocinasa es producida como el precursor de la prourocinasa por las células epiteliales que recubren los conductos excretorios. El papel de la urocinasa es activar la disolución de los coágulos de fibrina que pueden depositarse en estos conductos.
La tPA activa cliva al plasminógeno para producir plasmina la cual digiere a la fibrina; esta degradación resulta en un producto soluble al cual ni la plasmina ni el plasminógeno se pueden unir. Luego de la liberación del plasminógeno y la plasmina, éstos se inactivan rápidamente por sus respectivos inhibidores. La inhibición de la actividad de la tPA resulta de la unión de específicas proteínas inhibidoras. Por lo menos 4 diferentes inhibidores han sido identificados, dos de los cuales son inhibidores de los activadores del plasminógeno tipo I (PAI-1) y tipo 2 (PAI-2) tienen la mayor importancia fisiológica.
Activación plaquetaria Un enorme número de sustancias puede actuar sobre las plaquetas e inducir su activación [el factor de activación plaquetaria,6 trombina, el tromboxano (TXA2), fosfato de adenosina (ADP),10 epinefrina, vasopresina y serotonina] a través de sus respectivos receptores, pero todas lo hacen a merced de rutas de señalización intracelular que involucran un sistema de proteínas G acoplado a enzimas efectoras comolas fofolipasas A2 (PLA2) y C (PLC) y adenilato y guanilato ciclasas.5, 6, 10 Hasta ahora se han identificado nueve componentes diferentes de la familia de proteínas G en las plaquetas y, dependiendo del tipo de proteína G que participe en la señalización. Se puede activar o inhibir el efecto.6 Un ejemplo lo constituye la regulación por proteína G de la adenilato ciclasa y por tanto de la producción de monofosfato de adenosina cíclico (AMPc). Un incremento de las cantidades de AMPc bloquea la liberación de calcio en el STD e inhibe la función de la plaqueta. Este es el mecanismo que utilizan las prostaciclinas del endotelio vascular sano para inhibir la activación plaquetaria. DIAPOSITIVA #DOS (2) Coagulación de la Sangre Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre. Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional. El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse. Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas. Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
Coagulación de la sangre El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc. En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares. A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
Vía Intrínseca Recibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso hemostático. El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular. En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación. En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como la celulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción. A esta vía es posible subdividirla en tres pasos: -Formación del factor XIa En esta etapa participan cuatro proteínas: Precalicreína, Quininógeno de alto peso molecular (HMWK) y los factores XII y XI. Esta etapa no requiere de iones calcio. Estos cuatro factores se adsorben sobre la superficie cargada negativamente, formando el complejo cebador o de iniciación. De estos factores el XII funciona como verdadero iniciador, ya que si bien es una proenzima, posee una pequeña actividad catalítica que alcanza para activar a la precalicreína convirtiéndola en calicreína. En segunda instancia la calicreína actúa catalíticamente sobre el factor XII para convertirlo en XIIa, una enzima muchísimo más activa. La actividad catalítica de la calicreína se ve potenciada por el HMWK. Por último la proteasa XIIa actúa sobre el factor XI para liberar XIa. -Formación del factor IXa El factor IX se encuentra en el plasma como una proenzima. En presencia de iones Ca2+ el factor XIa cataliza la ruptura de una unión peptídica en la molécula del factor IX para formar un glucopéptido de 10 KDa y liberar por otro lado al factor IXa. El factor IX se encuentra ausente en personas con hemofilia tipo B. -Formación del factor Xa Sobre la membrana de las plaquetas se forma un complejo constituido por los factores IXa, X y VIII. Los residuos gamma-carboxiglutamato de los factores IXa y X actúan como quelantes del ion Ca2+, permitiendo que estos componentes formen un complejo unido por medio de puentes de iones calcio y ayudando a que el complejo se ancle a los fosfolípidos de membrana. Primero se unen los factores X y IXa a la membrana y luego se une el VIII. El factor VIII es en realidad un homorodímero, formado por cuatro cadenas proteicas, cada una codificada por un gen diferente (VIII:C y VIII:R). El componente VIII:C es conocido como "componente antihemofílico" y actúa como cofactor del IXa en la activación del factor X, el componente VIII:R es el que permite la unión del factor VIII al complejo. La ausencia del componente antihemofílico causa hemofilia A. El complejo formado por los factores IXa-X-VIII-Fosfolípidos y Ca2+ actúa sobre el factor X para convertirlo en Xa. En este punto concluye la vía intrínseca.
Trombo & Cuagulo Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida. Un coágulo sanguíneo que se forma dentro de una de las venas o las arterias se denomina trombo y también se puede formar en el corazón. Un trombo que se desprende y viaja desde un lugar en el cuerpo a otro se llama émbolo.Un trombo o émbolo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo. Una obstrucción en una arteria puede impedir que el oxígeno llegue a los tejidos en esa área, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede provocar daños en los tejidos o la muerte. Una obstrucción en una vena generalmente provocará acumulación de líquido e hinchazón. Las situaciones en las que es más probable que se forme un coágulo de sangre abarcan: Estar en reposo en cama por largo tiempo. Cruzar las piernas durante largos periodos al estar sentado o sentarse durante mucho tiempo, como en un avión o en un vehículo. Durante y después del embarazo. No tener suficiente agua en el cuerpo (deshidratación). Tomar píldoras anticonceptivas u hormonas estrógeno (especialmente en las mujeres que fuman). Utilizar un catéter intravenoso por largo tiempo. Los coágulos de sangre también son más probables en personas con cáncer, cirugía o lesión recientes, obesidad y enfermedades del hígado o del riñón. Una acumulación de colesterol que estreche una arteria puede cambiar o reducir el flujo de sangre, facilitando la formación de un coágulo sanguíneo o de un trombo. Las afecciones que se transmiten de padres a hijos (hereditarias) pueden hacerlo más propenso a la formación de coágulos sanguíneos anormales. Los trastornos hereditarios que afectan la coagulación son: Trombofilia por factor V de Leiden Mutación de la protrombina G20210A Otras afecciones raras como deficiencias de antitrombina III, proteína C y S Un coágulo de sangre puede bloquear una arteria o una vena en el corazón, afectando: El corazón (angina de pecho o un ataque cardíaco) Los intestinos (isquemia mesentérica) o trombosis venosa mesentérica Los riñones (trombosis de la vena renal) Las arterias de las piernas o brazos Las piernas (trombosis venosa profunda) Los pulmones (embolia pulmonar) El cuello o el cerebro (accidente cerebrovascular) Los trombos y émbolos pueden fijarse firmemente a un vaso sanguíneo y pueden bloquear parcial o completamente el flujo de sangre a dicho vaso. Un bloqueo en el vaso sanguíneo impide que el flujo normal de sangre y oxígeno llegue a los tejidos en ese sitio, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede ocasionar daño tisular o necrosis en esa área.
Fibrinólisis Después de que el coágulo se ha establecido, comienza la reparación de los tejidos afectados con el proceso de cicatrización. Para hacer posible esto el coágulo es colonizado por células que formarán nuevos tejidos y en el proceso va siendo degradado. La degradación de la fibrina (fibrinólisis), componente mayoritaria del coágulo, es catalizada por la enzima plasmina, una serina proteasa que ataca las uniones peptídicas en la región triple hélice de los monómeros de fibrina. La plasmina se genera a partir del plasminógeno, un precursor inactivo; activándose tanto por la acción de factores intrínsecos (propios de la cascada de coagulación) como extrínsecos, el más importante de los cuales es producido por el endotelio vascular. Se le denomina "activador tisular del plasminógeno" (t-PA). El gen de este factor ha sido clonado y actualmente se puede obtener la proteína producida por tecnología de ADN recombinante. Este factor suele utilizarse en clínica para favorecer la disolución de trombos. DIAPOSITIVA #OCHO (8) Sistema Fibrinolitico La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón. La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca: En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA. En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
La fibrinólisis es un proceso que previene los coágulos de sangre de crecimiento y convertirse en problemática [1] Este proceso tiene dos tipos:. Fibrinolisis primaria y fibrinólisis secundaria. El tipo principal es un proceso normal del cuerpo, mientras que la fibrinólisis secundaria es la descomposición de los coágulos debido a un medicamento, un trastorno médico, o alguna otra causa.
En la fibrinólisis, un coágulo de fibrina, el producto de la coagulación, se descompone. [2] Su plasmina principal enzima corta la malla de fibrina en varios lugares, que conduce a la producción de fragmentos circulantes que se eliminan por otras proteasas o por el riñón y el hígado .
La plasmina se produce en una forma inactiva, el plasminógeno, en el hígado. Aunque plasminógeno no puede escindir la fibrina, que todavía tiene una afinidad por ella, y se incorpora en el coágulo cuando se formó.
Activador del plasminógeno tisular (t-PA) [3] y uroquinasa son los agentes que convierten el plasminógeno en la plasmina activa, permitiendo de este modo que se produzca la fibrinólisis. t-PA se libera en la sangre muy lentamente por el endotelio dañado de los vasos sanguíneos, de tal manera que, después de varios días (cuando el sangrado se ha detenido), el coágulo se descompone. Esto ocurre porque plasminógeno se convirtió en atrapado dentro del coágulo cuando se formó, ya que se activa lentamente, se descompone la malla de fibrina. t-PA y uroquinasa se sí mismos inhibidas por el inhibidor del activador del plasminógeno-1 y el inhibidor del activador del plasminógeno-2 (PAI-1 y PAI-2). En contraste, la plasmina estimula aún más la generación de plasmina mediante la producción de formas más activas de tanto activador del plasminógeno tisular (tPA) y la uroquinasa.
Alfa 2-antiplasmina y alfa 2-macroglobulina inactivan plasmina. Actividad de la plasmina también se reduce por el inhibidor de la fibrinólisis activable por trombina (TAFI), que modifica la fibrina para que sea más resistente a la del plasminógeno mediada por tPA.
Massiel Rosario 89427. Activacion Plaquetaria: Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro,1 derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos. Las plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular; Estos dos factores de crecimiento han demostrado jugar un papel significativo en la regeneración y reparación del tejido conectivo; Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.3 4 5 6 7 8 9. La función plaquetaria consiste en el mantenimiento del corazón; Esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio. La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria(plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación. Las plaquetas activadas cambian su forma haciéndose más esféricas, y formando pseudopodos en su superficie. De esta forma toman una forma estrellada.
La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP.
Las células endoteliales producen una proteína llamada factor de von Willebrand (FvW),un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas. Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo. Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio. La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria(plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre. Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional. El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse. Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas. Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
Factores de coagulación
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc. En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares. A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos. Siete de los factores de coagulación (precalicreína —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas. Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada. Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola. Algunos factores de coagulación requieren vitamina K para su síntesis en el hígado, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
1. La coagulación comienza en respuesta a una lesión en un vaso sanguíneo. Es un proceso donde se producen una serie de reacciones en cadena de varios tipos celulares y proteínas para formar un coágulo evitando asi la pérdida excesiva de sangre. Un coágulo consiste en una red de proteínas insolubles como la fibrina con plaquetas y células atrapadas que bloquea la salida de sangre hasta que se repare el tejido.
2. En la fase de iniciación es fundamental la formación de pequeñas cantidades de trombina. La lesión expone las proteínas solubles de la sangre a tejidos circundantes y a la matriz extracelular. Estos genera una serie de reacciones que tiene lugar cuando el factor VIIa se une a los factores tisulares en las membranas de las células que están fuera de los vasos originando el complejo VIIa/TF. Activa a su vez pequeñas cantidades de factor IX y X. El factor Xa se asocia con el factor Va en la superficie de las células que presentan los factores tisulares.
3. Se forma un complejo protrombinasa que da origen a pequeñas cantidades de trombina (factor IIa). El factor V puede ser activado por factor Xa, por proteasas de la matriz extracelular o ser liberado de las plaquetas al contactar con el colágeno u otros componentes de la matriz extracelular.
En la fase de amplificación se produce la activación de las plaquetas por la trombina generada en la fase de iniciación. Factores en la superficie de las plaquetas provocan la producción masiva de trombina. Tiene lugar sólo si la lesión permite la salida de plaquetas y del complejo VIII/vWF. Se secretan formas parcialmente activadas de factor V en la superficie. Se disocia el complejo VIII/vWF, permitiendo al factor vWF mediar la adhesión y agregación adicional de plaquetas y al factor VIII activarse y unirse a la membrana de las plaquetas. La trombina activa el factor XI en la superficie de la plaqueta durante esta fase.
En la fase de propagación se produce trombina de forma masiva para la formación de un coágulo estable. El factor IXa, generado en la fase de iniciación, se une al factor VIIIa en la superficie de las plaquetas. El factor XIa en la superficie de plaquetas permite la unión de más cantidad de factor IXa. El factor Xa generado por el complejo IXa/VIIIa formado en la membrana de las plaquetas se une con el factor Va. Al ensamblarse la protrombinasa (Xa/Va) provoca una masiva producción de trombina en cantidad suficiente para que se forme el coágulo.



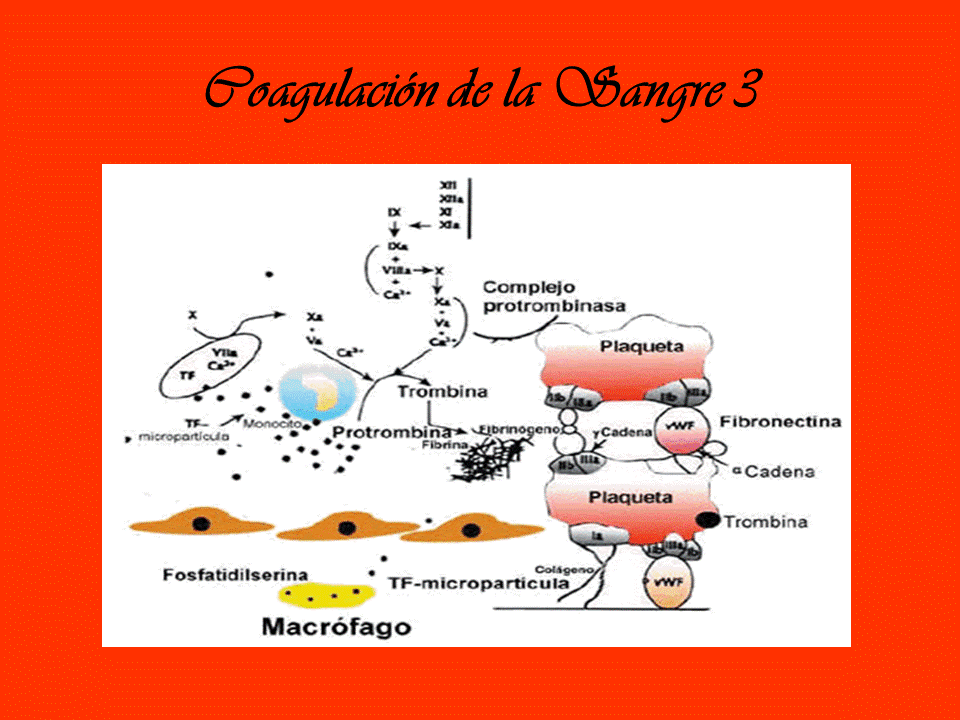




Plaquetas, cuagulacion y fibrinolisis por Juan F. Martinez 84445
ResponderEliminar-Activacion Plaquetaria:
Esta primera imagen nos muestra como se lleva a cabo activacion plaquetaria:
-Primero tiene que haber una lesion, donde el factor vW procede a adherirse al colageno expuesto en la lesion endotelial.
- segundo la adhesion: las plaquetas se adhieren al factor vW via el receptor GpIb en el lugar de la lesion unicamente. Las plaquetas comienzan a liberar ADP y Calcio necesario para la cascada de cuagulacion. El ADP ayuda a que las plaquetas se adhieran al endotelio.
-tercero la activacion: el ADP enlazado al receptor induce la expresion de GpIIb/IIIa en la superficie de las plaquetas.
-cuarto: agregacion: el fibrinogeno se adhiere al receptor GpIIb/ IIIa y enlazan plaquetas.
-Coagulacion:
En cuanto a la cuagulacion sabemos que esta tiene dos vias la via intrinseca y la via extrinseca. La via extrinseca consiste de los factores VII que es activado por el factor tisular para formar VIIa que a su vez escinde al factor X en Xa, que a su vez va a escindir el factor II (protrombina) en IIa (trombina) que a su vez escinde al fibrinogeno para formar los monomeros de fibrina que forman la red de fibrina que actua para estabilizar el tapon de plaquetas. Por otra parte la via intrinseca consta del factor XII que es activado por colageno, membrana basal o plaquetas activadas y escindido a XIIa que a su vez escinde al factor IX en IXa que a su vez es escindido por el factor VIIIa y de esta forma el factor IXa escinde al factor X y se conectan ambas vias para obtener el mismo resultado la red de fibrina y lo que es la cuagulacion.
-Fibrinolisis y sistema fibrinolitico:
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. Este diagrama nos resume y nos dice que esta se puede llevar a cabo por diversas vias. La activación de plasmina a partir de plasminógeno puede ocurrir a través de las vías, extrínseca, intrínseca, exogena y endogena: En la vía extrínseca se produce una segregación de diversas sustancias que posibilitan la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA. En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno que activa la plasmina y degradan los productos de la fibrina. No esta demás mencionar que la antitrombina inhibe las formas activadas de los factores II, VII, IX, X, XI y XII.
Bielka M. Nuñez 85699
ResponderEliminarEl proceso de agregación plaquetaría se desarrolla en dos secuencias:
En primer lugar, mediante la adhesión de las plaquetas a la superficie vascular lesionada (subendotelio) y posteriormente, mediante la activación plaquetaría, cuyo resultado final consiste en la formación de un entramado de plaquetas y cadenas de fibrinogeno que forma el trombo.
La activación plaquetaria comprende:
-la formación y liberación de sustancias vasoactivas o agonistas (tromboxano A2, adenosina trifosfato, trombina,epinefrina, serotonina y colageno) que favorecen al proceso de agragacion induciendo a su vez la activacion de otras plaquetas mediante una reaccion de cascada.
- la activacion de receptores de proteinas en la membrana plaquetaria, entre los que se encuentra el responsable de la fijación de las plaquetas a la zona lesionada (factor de von willerbrand) y el mas importante, el receptor glucoproteinas IIb\IIIa que reconoce y fija las cadenas de fibrinogeno, formando la trama final del tapon hemostatico.
-Factor de la coagulación I, Se convierte en fibrina por acción de la trombina. La fibrina constituye la red que forma el coágulo.en el plasma xiste una cantidad de 250-400mg\dl.
-Factor de coagulación II tambien conocido como prototrombina. Se convierte en trombina por la acción del factor Xa. La trombina cataliza la formación de fibrina a partir de fibrinógeno.en el plasma existe en una cantidad de 10-14mg\dl.
-factor III o factor tisular de trmboplastina. Se libera con el daño celular; participa junto con el factor VIIa en la activación del factor X por la vía extrínseca.
-factor IV su nombre es ion calcio,
Media la unión de los factores IX, X, VII y II a fosfolípidos de membrana. Su nivel en el plasma es de 4-5mg\dl.
El trombo es un coágulo sanguíneo que se forma en un vaso y permanece allí. La embolia es un coágulo que se desplaza desde el sitio donde se formó a otro lugar en el cuerpo. El trombo o embolia puede producirse en un vaso sanguíneo y obstruir el flujo sanguíneo en ese lugar, impidiendo el suministro de oxígeno y flujo sanguíneo a los tejidos circundantes. Esto puede ocasionar un daño, destrucción (infarto) e incluso la muerte o necrosis de los tejidos que se encuentran en esa área.
Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida.
Un coágulo sanguíneo que se forma dentro de una de las venas o las arterias se denomina trombo y también se puede formar en el corazón.
Un trombo que se desprende y viaja desde un lugar en el cuerpo a otro se llama émbolo.
Un trombo o émbolo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo.
Una obstrucción en una arteria puede impedir que el oxígeno llegue a los tejidos en esa área, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede provocar daños en los tejidos o la muerte.
Una obstrucción en una vena generalmente provocará acumulación de líquido e hinchazón.
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos.
La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
La fibrinolisis es un encadenamiento (proceso) enzimático compuesto por una serie de activadores e inhibidores los cuales regulan la conversión del proenzima circulante, plasminógeno, en el enzima activo plasmina. La producción de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina y esto es muy importante para el mantenimiento de la permeabilidad vascular . los componentes de la fibrinolisis son: plasminogeno, plasmita, activador tisular del plasminogeno, estreptoquinasa, uroquinasa, factor XII, precalicreina y quiminogeno de alto PM, PAI, glicoproteina rica en histidina, antiplasminas y a 2-antiplasmina.
Ana M. Igartua 86978
ResponderEliminarActivacion plaquetaria
La sangre circula por el interior de los vasos, los glóbulos rojos (la más abundante masa celular sanguínea) lo hace por el centro del vaso, mientras que las plaquetas circulan más próximas a la superficie endotelial para vigilar la integridad del endotelio. El endotelio normal proporciona una superficie no adhesiva para las plaquetas. Cuando se rompe el endotelio, el colágeno expuesto favorece la adhesión de las plaquetas por medio de la interacción con una cantidad de receptores, como vemos en el video, pero es esencial para concretar la adhesión el Factor de Von Willebrand. A partir de esto y con el aumento del Ca++ intracelular se produce la activación de las plaquetas, lo que genera una serie de eventos que favorece la agregación plaquetaria, como son:
-Cambio de forma y de ser una esfera regular pasa a tener prolongaciones.
-Liberación del contenido de los gránulos (degranulación):
ADP que torna a las plaquetas pegajosas,TX A2 derivado del Ácido Araquidónico (vasoconstrictor y agreganteplaquetario)
actor II (trombina).
Adhesión, Activación y Agregación son las fases que participan en la formación del tapón plaquetario .
Proceso de coagulacion
Los factores de coagulación son proteínas de la sangre que controlan el sangrado.
Cuando un vaso sanguíneo se lesiona, sus paredes se contraen para limitar el flujo de sangre al área dañada. Entonces, pequeñas células llamadas plaquetas se adhieren al sitio de la lesión y se distribuyen a lo largo de la superficie del vaso sanguíneo. Al mismo tiempo, pequeños sacos al interior de las plaquetas liberan señales químicas para atraer a otras células al área y hacer que se aglutinen a fin de formar lo que se conoce como tapón plaquetario.
En la superficie de estas plaquetas activadas muchos factores de coagulación diferentes trabajan juntos en una serie de reacciones químicas complejas (conocidas como cascada de la coagulación) para formar un coágulo de fibrina. El coágulo funciona como una red para detener el sangrado.
Los factores de la coagulación circulan en la sangre sin estar activados. Cuando un vaso sanguíneo sufre una lesión se inicia la cascada de la coagulación y cada factor de la coagulación se activa en un orden específico para dar lugar a la formación del coágulo sanguíneo. Los factores de la coagulación se identifican con números romanos (e. g. factor I o FI).
Fibrinolisis
La fibrinólisis primaria se refiere a la descomposición normal de los coágulos, mientras que la fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa.
Consecuencias
Cuando se produce demasiado rápidamente, puede provocar hemorragias espectaculares (fibrinólisis hemorrágica) intra o postoperatorias (particularmente en Cirugía Torácica), o después de un parto, de un aborto o de un choque traumático; o bien puede provocar hemorragias menos importantes (hematuria) en el curso de ciertos cánceres (próstata, páncreas, estómago), de las cirrosis y de las leucosis. Obedece a la liberación de activadores de la profibrinolisina, o bien a la producción de un fermento proteolítico por los tejidos enfermos.
Activación plaquetaria
ResponderEliminarEl proceso de formación del trombo es la consecuencia patológica de la activación del mecanismo hemostático, el cual depende de un sistema enzimático complejo regulado por la acción de diversos factores activadores e inhibidores. En su desarrollo están implicados los elementos circulantes, las estructuras de la pared vascular y las proteínas plasmáticas.
El proceso de agregación plaquetaria se desarrolla en dos secuencias: en primer lugar, mediante la adhesión de las plaquetas a la superficie vascular lesionada (subendotelio); y, posteriormente, mediante la activación plaquetaria, cuyo resultado final consiste en la formación de un entramado de plaquetas y cadenas de fibrinógeno que forman el trombo.
Por su parte, la activación de las plaquetas comprende:
a) la formación y liberación de sustancias vasoactivas o agonistas plaquetarios (tromboxano A2, adenosina difosfato, trombina, epinefrina, serotonina, y colágeno) que favorecen el proceso de agregación induciendo a su vez la activación de otras plaquetas mediante una reacción en cascada
b) la activación de receptores de proteínas en la membrana plaquetaria, entre los que se encuentra el responsable de la fijación de las plaquetas a la zona lesionada (factor de Von Willebrand) y el más importante, el receptor glucoproteína IIb/IIIa que reconoce y fija las cadenas de fibrinógeno, formando la trama final del tapón hemostático.
Fisiológicamente, todo este proceso puede ser a su vez inhibido por otras sustancias, principalmente prostaciclina y óxido nítrico, que aumentan las concentraciones de AMP-cíclico en las plaquetas y en la pared vascular mediante una estimulación de la adenilciclasa (prostaciclina) o bien, aumentan las concentraciones de GMP-cíclico por estímulo de la guanilciclasa (óxido nítrico)
Coagulación de la sangre 1
ResponderEliminarCuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden lavasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimáticopor el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse.
Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas.
Coagulación de la sangre 2
ResponderEliminarEl proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc.
En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares.
A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
Coagulación de la sangre 3
ResponderEliminarLa cascada de coagulación se divide para su estudio, clásicamente en tres vias: La via intrínseca, la vía extrínseca y la vía común.
Las vías intrínseca y extrínseca son las vías de iniciación de la cascada, mientras que la vía común es hacia donde confluyen las otras dos desembocando en la conversión de fibrinógeno en fibrina.
Esta división es un tanto arbitraria y tiene más que ver con las deficiencias de las técnicas que en su momento se utilizaron para desentrañar los mecanismos implicados, que con lo que ocurre realmente en una lesión vascular; ya que en este último caso se establecen varias interrelacciones entre las vías de iniciación.
Cada reacción de estas vías da como resultado el ensamblado de un complejo compuesto por una enzima (factor de coagulación activado), un sustrato (proenzima de un factor de coagulación) y un cofactor que actúa posibilitando la reacción.
Estos componentes se ensamblan en general sobre una superficie fosfolipídica y se mantienen unidos por medio de puentes formados por iones Ca2+. Por lo tanto la reacción en cascada tiende a producirse en un sitio donde este ensamblaje puede ocurrir; por ejemplo sobre la superficie de plaquetas activadas.
Tanto la vía intrínseca como la vía extrínseca desembocan en la conversión del factor X en Xa (la letra "a" como subíndice "a" significa "activado") punto en el que se inicia la vía común.
Coagulación de la sangre 4
ResponderEliminarRecibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso hemostático.
El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular.
En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación.
En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como la celulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción.
A esta vía es posible subdividirla en tres pasos:
Recibió este nombre debido a que fue posible notar desde un primer momento que la iniciación de esta vía requería de factores ajenos a la sangre.
Cuando la sangre entra en contacto con tejidos lesionados o se mezcla con extractos de tejidos, se genera muy rápidamente factor Xa. En este caso la activación de la proenzima X es mediada por un complejo formado por factor VII, Ca2+ y factor tisular unido a fosfolípidos provenientes de las membranas celulares rotas y de las plaquetas (antiguamente este complejo factor tisular-fosfolípidos era conocido como tromboplastina).
El factor tisular es una lipoproteína sintetizada en el endotelio de los vasos sanguíneos de todos los tejidos, aunque es especialmente abundante en pulmón, cerebro y placenta. El factor tisular se encuentra normalmente "secuestrado" en el interior de las células endoteliales y es secretado en respuesta a una lesión, o bajo el efecto de algunas citoquinas tales como el Factor de Necrosis Tumoral (TNF), InterLeucina 1 (IL-1); o por endotoxinas bacterianas.
La vía extrínseca es muy rápida, se cumple en apenas unos segundos y comprende dos pasos; mientras que la intrínseca insume varios minutos.
Coagulo y trombo
ResponderEliminarTrombo: es una masa intravascular o intracardíaca formada por elementos de la sangre e insolubles en ella, se caracterizan por estar fijos a la pared del vaso por una zona donde son más rígidos y consistentes. Pueden ser arteriales o venosos.
Coagulos: intravasculares son postmorten son gelatinosos, con una porción declive rojo oscura porque los eritrocitos sedimentan por acción de la graverdad y un sobrenadante amarillo en grasa de pollo, no estan fijos a la pared del vaso. En cambio los trombos son mas consistentes, estan anclados en un punto a la pared del vaso o cardiaca, los arteriales consistentes de color blanco grisaceo y friables, formados por una red de plaquetas fibrina y leucocitos en degeneración (se generan en zonas donde hay turbulencia, como en la bifircación de los vasos) lo venosos son rojos y se desarrollan en zonas de estasis sanguineo por lo que retienen mayor número de hematíes, por eso son rojos
Fibrinolisis
ResponderEliminarLa fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos.
La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón.
La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca:
• En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA.
• En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno.
La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y elinhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
El sistema fibrinolítico
ResponderEliminarEl sistema fibrinolítico va a jugar además un papel importante en diversas situaciones en las que se produce proteolisis tisular, tales como inflamación, invasión tumoral o neovascularización (3, 4). La importancia fisiopatológica del sistema fibrinolítico deriva de que las alteraciones que condicionan un defecto de actividad fibrinolítica pueden predisponer a la trombosis, mientras que un exceso de activación favorecería la aparición de hemorragia. Además va a tener una gran importancia en el área terapéutica por la aplicación creciente de fármacos fibrinolíticos en el tratamiento de la trombosis, especialmente en el infarto agudo de miocardio (IAM).
Principales componentes del sistema fibrinolítico: La fibrinolisis es un encadenamiento (proceso) enzimático compuesto por una serie de activadores e inhibidores los cuales regulan la conversión del proenzima circulante, plasminógeno, en el enzima activo plasmina. La producción de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina (5) y esto es muy importante para el mantenimiento de la permeabilidad vascular
El plasminógeno o profibrinolisina es una glicoproteína sintetizada por el hígado, presente en el plasma sanguíneo y la mayor parte del fluido extracelular como el precursor inactivo de una enzima proteasa llamada plasmina. El plasminógeno es el componente central del sistema fibrinolítico del organismo, es decir, en la disolución de coágulos sanguíneos de craneados y otros organismos eucariotas. En humanos, la concentración de plasminógeno es entre 1.5 a 2 µmol/l.
Josue O. Valdes 87949
ResponderEliminarLas plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro,1 derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos.
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo. La función plaquetaria consiste en el mantenimiento del corazón; esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio.
ResponderEliminarLa Activación plaquetaria consiste cuando la superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP.
La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria(plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
El proceso de coagulación implica una serie de reacciones que intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares. A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (preacelerina factor V, protrombina Factor II, proconvertina factor VII, factor antihemofílico beta IX, factor Stuart X, tromboplastina plasmática XI y factor Hageman XII) son zimógenos sintetizados en el hígado.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada. Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
El fibrinógeno (factor I) es una glicoproteína compuesta por seis cadenas polipeptídicas: dos A-alfa, dos B-beta y dos gamma; unidas entre sí por puentes disulfuro.
Durante la fibrinolisis, después de que el coágulo se ha establecido, comienza la reparación de los tejidos afectados con el proceso de cicatrización. Para hacer posible esto el coágulo es colonizado por células que formarán nuevos tejidos y en el proceso va siendo degradado.
La degradación de la fibrina (fibrinólisis), componente mayoritaria del coágulo, es catalizada por la enzima plasmina, una serina proteasa que ataca las uniones peptídicas en la región triple hélice de los monómeros de fibrina.
La plasmina se genera a partir del plasminógeno, un precursor inactivo; activándose tanto por la acción de factores intrínsecos (propios de la cascada de coagulación) como extrínsecos, el más importante de los cuales es producido por el endotelio vascular. Se le denomina "activador tisular del plasminógeno" (t-PA).
El gen de este factor ha sido clonado y actualmente se puede obtener la proteína producida por tecnología de ADN recombinante.
Este factor suele utilizarse en clínica para favorecer la disolución de trombos.
Zoraya Perozo 85791
ResponderEliminarEl sistema de la coagulación mantiene la integridad vascular limitando la hemorragia y remodelando el vaso dañado, una vez que los mecanismos de reparación tisular han cesado. La hemostasia es el conjunto de mecanismos fisiológicos que se encargan de detener la hemorragia cuando se produce una injuria o daño vascular, es decir que existe un equilibrio en la necesidad fisiológica de mantener la sangre líquida permitiendo su circulación y cumplir sus funciones de transporte de oxígeno y elementos nutritivos a los tejidos y por otra parte la capacidad de detener el flujo cuando ocurre injuria vascular.
¿Quiénes conforman el sistema hemostático?
1- Vasos sanguíneos
2- Plaquetas
3- Mecanismo de la coagulación plasmática
4- Mecanismo fibrinolisis
5- Inhibidores o reguladores fisiológicos
¿Cuál es la secuencia cuando se rompe un vaso sanguíneo?
1. reacción o fase vascular (modificadores del tono vascular)
2. Reacción o fase plaquetaria (adhesión y agregación plaquetaria con la formación del trombo hemostático primario)
3. Activación de la coagulación sanguínea (trombo hemostático definitivo)
4. Activación de la fibrinolisis (digestión del trombo, recanalización del vaso)
EQUILIBRIO ENTRE LA COAGULACIÓN Y FIBRINÓLISIS
La trombina tiene un efecto anticoagulante local, al conformar un complejo con la trombomodulina sobre la superficie endotelial, inhibe el coágulo sanguíneo por activación de la proteína C. La proteína C (proteína k dependiente), inicia la fibrinólisis por activación del plasminógeno y neutralizando un inhibidos activador del plasminógeno (PAI-1).
Las antitrombinas son inhibidores de la trombina y otras proteasas de la coagulación. La Antitrombina III es el más importante y posiblemente el causante del 50% de la fluidez natural de la sangre. La acción de la antitrombina III es catalizada por la heparina y por otras enzimas de la cascada de la coagulación como los factores Xa, XII, XI y X. Una deficiencia de antitrombina III se encuentra asociada a un aumento de trombosis.
La trombina tiene un efecto anticoagulante local, al conformar un complejo con la trombomodulina sobre la superficie endotelial, inhibe el coágulo sanguíneo por activación de la proteína C. La proteína K dependiente), inicial la fibrinólisis por activación del plasminógeno y neutralizando un inhibidor del plasminógeno (PAI-1)
FORMACIÓN DE FIBRINA Y FIBRINÓLISIS
La introducción de la terapia a base de t-PA (tejido activador del plasminógeno) en la práctica clínica, ha puesto de presente la importancia de la fibrinólisis en el mantenimiento y función de la pared arterial. La formación de fibrina es regulada por un proceso enzimático de degradación que convierte la fibrina en fibrinógeno por acción de la plasmina. La plasmina está formada a partir de plasminógeno una globulina-b sintetizada por el hígado. La actividad de la plasmina está determinada por activadores o inhibidores del plasminógeno o inhibidores de la plasmina. La fibrinólisis es iniciada por el factor XIIA o uroquinasa activadora de plasminógeno (vía intríseca) o por un activador del plasminógeno (t-PA) vía intrínseca.
Los estrógenos disminuyen una glicoproteína rica en histidina y puede aumentar la fibrinólisis. Las progestinas están asociadas con una aumento en la actividad del plasminógeno.
Clínicamente como marcadores de trombosis se pueden utilizar el fibrinógeno plasmático, el tiempo de protrombina (indirectamente mide el factor VII) y el PAI-1. Recientemente niveles elevados de los fragmentos 1.2 (derivan de la conversión de protrombina o trombina). El PTT es de poco valor. El fibrinopéptido A y B así como varios productos de degradación del fibrinógeno, cambian en respuesta a la terapia hormonal y pueden medir indirectamente la aceleración de la coagulación. La dosificación de proteína C, S y posible a-antiplasmina, miden la acción de la Antitrombina III, en casos de deficiencia de esta proteínas puede ser útil para prevención de la trombosis, pero no informa sobre el riesgo de la inapropiada formación del trombo.
youel dias 86760
ResponderEliminartrombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro,1 derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos.
Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva. Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebro vascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores. Cualquier anormalidad o enfermedad de las plaquetas se denomina trombocitopatía,2 la cual puede consistir, ya sea en tener un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Se pueden producir desórdenes que reducen el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos. Sin embargo, otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragias.
Las plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular; Estos dos factores de crecimiento han demostrado jugar un papel significativo en la regeneración y reparación del tejido conectivo; Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.
youel diaz 86760
ResponderEliminartrombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro,1 derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos.
Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva. Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebro vascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores. Cualquier anormalidad o enfermedad de las plaquetas se denomina trombocitopatía,2 la cual puede consistir, ya sea en tener un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Se pueden producir desórdenes que reducen el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos. Sin embargo, otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragias.
Las plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular; Estos dos factores de crecimiento han demostrado jugar un papel significativo en la regeneración y reparación del tejido conectivo; Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.
youel diaz 86760
ResponderEliminarLas plaquetas son producidas en el proceso de formación de las células sanguíneas llamado (trombopoyesis) en la médula ósea, por fragmentación en los bordes citoplasmáticos de los megacariocitos.
El rango fisiológico de las plaquetas es de 150-400 x 109/litro.
Alrededor de 1 x 1011 plaquetas de media son producidas cada día por un adulto sano.
La expectativa de vida de las plaquetas circulantes es de 7 a 10 días.
La producción de megacariocitos y plaquetas está regulada por la trombopoyetina, una hormona producida usualmente por el hígado y los riñones.
Cada megacariocito produce entre 5.000 y 10.000 plaquetas.
Las plaquetas son destruidas por fagocitosis en el bazo y por las células de Kupffer en el hígado.
Una reserva de plaquetas es almacenada en el bazo y son liberadas cuando se necesitan por medio de contracción esplénica mediada por el sistema nervioso simpático.
Formación de trombos
La función plaquetaria consiste en el mantenimiento de la hemostasia; Esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio.
Activación
La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP. Las células endoteliles producen una proteína llamada factor de von Willebrand (FvW),un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas. Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo. Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio. La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria; estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
Cambio de forma
ResponderEliminarLas plaquetas activadas cambian su forma haciéndose más esféricas, y formando pseudopodos en su superficie. De esta forma toman una forma estrellada.
Secreción de gránulos
Las plaquetas contienen gránulos alfa y gránulos densos. Las plaquetas activadas excretan el contenido de estos gránulos dentro de sus sistemas canaliculares y en la sangre circundante. Existen dos tipos de gránulos:
Gránulos densos (contienen ADP o ATP, calcio, y serotonina)
Granulos-α (contienen factor 4 plaquetario, factor de crecimiento transformante beta 1 (TGF beta 1), factor de crecimiento derivado de plaquetas, fibronectina, B-tromboglobulina, FvW, fibrinógeno, y factores de coagulación factor V y XIII).
Síntesis de tromboxano A2
La activación plaquetaria inicia la vía del ácido araquidónico para producir Tromboxano A2; el tromboxano A2 está involucrado en la activación de otras plaquetas y su formación es inhibida por los inhibidores de la COX, como el ácido acetilsalicílico. [editar] Adhesión y agregación La agregación plaquetaria, usa el fibrinógeno y el FvW como agentes conectores. El receptor de agregación plaquetaria más abundante es la glicoproteina IIb/IIIa (gpIIb/IIIa); se trata de un receptor para el fibrinógeno dependiente del calcio, fibronectina, vitronectina, trombospondina, y factor de von Willebrand (FvW). Otros receptores incluyen el complejo GPIb-V-IX (FvW) y GPVI (colágeno).ó Las plaquetas activadas se adherirán, via glicoproteína (GP) Ia, al colágeno expuesto por el daño epitelial. La agregación y adhesión actúan juntos para formar el tapón plaquetario. Los filamentos de Miosina y actina en las plaquetas son estimuladas para contraerse durante la agregación, reforzando todavía más el tapón. La agregación plaquetaria es estimulada por el ADP, tromboxano, y la activación del receptor-α2, pero inhibido por agentes antiinflamatorios como las prostaglandinas PGI2 y PGD2. La agregación plaquetaria se ve aumentada por la administración exógena de esteroídes anabólicos.
Reparación de heridas
El coágulo sanguíneo es solo una solución temporal para detener la hemorragia; la reparación del vaso debe ocurrir después. La agregación plaquetaria ayuda en este proceso mediante la secreción de sustancias químicas que promueven la invasión de fibroblastos del tejido conectivo adyacente hacia el interior de la herida para formar una costra. El coágulo obturador es lentamente disuelto por la enzima fibrinolítica, plasmina, y las plaquetas son eliminadas por fagocitosis.
Otras funciones
Retracción del coágulo
Pro-coagulación
Inflamación
Señalización citoquínica
Fagocitosis
Señalización citoquínica
Adicionalmente a su función de ser el efector celular de la hemostasia, las plaquetas son rápidamente depositadas en sitios de lesión o infección, y potencialmente modulan los procesos inflamatorios por medio de la interacción con leucocitos y por la secreción de citoquinas, quimiosinas, y otros mediadores de la inflamación. Las plaquetas también secretan factor de crecimiento derivado de plaquetas (PDGF).
Papel en enfermedades
ResponderEliminarRecuentos altos y bajos
El recuento de plaquetas de un individuo sano se encuentra entre 150,000 y 450,000 por μl (microlitro) de sangre (150-450 x 109/L).18 El 95 % de los individuos sanos tendrán recuentos de plaquetas dentro de este rango. Algunos tendrán recuentos de plaquetas estadísticamente anormales sin tener ninguna anormalidad demostrable. Sin embargo, si el recuento es muy alto o muy bajo la probabilidad de que una anormalidad este presente es más alta. Tanto la trombocitopenia como la trombocitosis pueden manifestarse como problemas de coagulación. En general, los recuentos bajos de plaquetas incrementan el riesgo de sangrado; sin embargo existen excepciones. Por ejemplo la trombocitopenia inmune inducida por heparina. En la trombocitosis se puede producir trombosis, sin embargo esto sucede principalmente cuando el recuento elevado es debido a desordenes mieloproliferativos. Los recuentos de plaquetas en general, no son corregidos con transfusión a menos que el paciente esté sangrando o el recuento haya caído por debajo 5 x 109/L. La transfusión esta contraindicada en la púrpura trombocitopénica idiopática (PTI), puesto que estimula la coagulopatía. En los pacientes sometidos a cirugía, niveles inferiores a 50 x 109/L están asociados a sangrado quirúrgico anormal, y procedimientos anestésicos regionales como la anestesia epidural son evitados para niveles inferiores a 80-100 x 109/L. El recuento normal de plaquetas no es garantía de función adecuada. En algunos estados, las plaquetas, siendo normales en número, son disfuncionales. Por ejemplo, el ácido acetilsalicílico interrumpe irreversiblemente la función plaquetaria mediante la inhibición de la ciclooxigenasa-1 (COX1), y por consiguiente la hemostasia normal. Las plaquetas resultantes no tienen ADN y son incapaces de producir nueva ciclooxigenasa. La función plaquetaria normal no se restaurara hasta que el uso de ASA haya cesado y un número suficiente de las plaquetas afectadas hayan sido reemplazadas por nuevas, lo cual suele tardar unos 7 días. El Ibuprofeno, un AINE, no tiene un período tan largo de efecto, y la función plaquetaria vuelve a la normalidad dentro de las 24 horas,19 y tomando ibuprofeno antes que el ASA prevendrá los efectos irreversibles de esta.20 La Uremia, a consecuencia de la insuficiencia renal, conduce a la disfunción plaquetaria que puede ser aminorada con la administración de desmopresina.
youel diaz 86760
ResponderEliminarLa plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón.
La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca:
En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA.
En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno.
Maritza Gonzalez 88598
ResponderEliminarActivacion plaquetaria
El interior de los vasos sanguíneos está revestido por una capa de células endoteliales las cuales actúan inhibiendo la activación plaquetaria mediante la producción de ciertos factores. Además producen una proteína llamada factor de von Willebrand (vWF), el cual permite a las células endoteliales a adherir el colágeno a la membrana basal. Cuando la capa endotelial es lesionada, el colágeno, el vWF y el factor tisular del endotelio son expuestos al flujo sanguíneo. Las plaquetas hacen contacto con el colágeno o el vWF son activadas. La activación plaquetaria resulta en el transporte de fosfolípidos cargados a la superficie plaquetaria que proporcionan una superficie para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación. Las plaquetas se hacen más esféricas y forman pseudopodos en su superficie. Estas contienen gránulos alfa y gránulos densos, excretan el contenido de estos gránulos dentro de sus sistemas canaliculares y en la sangre circundante. Existen dos tipos de gránulos: Gránulos densos (contienen ADP o ATP, calcio, y serotonina) y Granulos-α (contienen factor 4 plaquetario, factor de crecimiento transformante beta 1 (TGF beta 1), factor de crecimiento derivado de plaquetas, fibronectina, B-tromboglobulina, FvW, fibrinógeno, y factores de coagulación factor V y XIII).
Maritza Gonzalez 88598
ResponderEliminarCoagulacion de la Sangre
1. La coagulación comienza en respuesta a una lesión en un vaso sanguíneo. Es un proceso donde se producen una serie de reacciones en cadena de varios tipos celulares y proteínas para formar un coágulo evitando asi la pérdida excesiva de sangre. Un coágulo consiste en una red de proteínas insolubles como la fibrina con plaquetas y células atrapadas que bloquea la salida de sangre hasta que se repare el tejido.
2. En la fase de iniciación es fundamental la formación de pequeñas cantidades de trombina. La lesión expone las proteínas solubles de la sangre a tejidos circundantes y a la matriz extracelular. Estos genera una serie de reacciones que tiene lugar cuando el factor VIIa se une a los factores tisulares en las membranas de las células que están fuera de los vasos originando el complejo VIIa/TF. Activa a su vez pequeñas cantidades de factor IX y X. El factor Xa se asocia con el factor Va en la superficie de las células que presentan los factores tisulares.
3. Se forma un complejo protrombinasa que da origen a pequeñas cantidades de trombina (factor IIa). El factor V puede ser activado por factor Xa, por proteasas de la matriz extracelular o ser liberado de las plaquetas al contactar con el colágeno u otros componentes de la matriz extracelular.
En la fase de amplificación se produce la activación de las plaquetas por la trombina generada en la fase de iniciación. Factores en la superficie de las plaquetas provocan la producción masiva de trombina. Tiene lugar sólo si la lesión permite la salida de plaquetas y del complejo VIII/vWF. Se secretan formas parcialmente activadas de factor V en la superficie. Se disocia el complejo VIII/vWF, permitiendo al factor vWF mediar la adhesión y agregación adicional de plaquetas y al factor VIII activarse y unirse a la membrana de las plaquetas. La trombina activa el factor XI en la superficie de la plaqueta durante esta fase.
En la fase de propagación se produce trombina de forma masiva para la formación de un coágulo estable. El factor IXa, generado en la fase de iniciación, se une al factor VIIIa en la superficie de las plaquetas. El factor XIa en la superficie de plaquetas permite la unión de más cantidad de factor IXa. El factor Xa generado por el complejo IXa/VIIIa formado en la membrana de las plaquetas se une con el factor Va. Al ensamblarse la protrombinasa (Xa/Va) provoca una masiva producción de trombina en cantidad suficiente para que se forme el coágulo.
4. Via Intrinseca: requiere de los factores de coagulación VIII, IX, X, XI y XII y de proteínas como la precalicreina (PK) y el quininógeno de alto peso molecular (HK o HMWK), al igual que iones de calcio y fosfolípidos secretados por las plaquetas. Los componentes resultan en la conversión del factor X al factor Xa. La iniciación de la vía intrínseca ocurre cuando la precalicreina, HMWK, factor XI y el factor XII son expuestos a una superficie de carga negativa, puede ocurrir como el resultado de una interacción con los fosfolípidos de las partículas lipoproteínas circulantes tales como los quilomicrones, VLDL y LDL oxidada.
5. Via Extrinseca: iniciada en el sitio de la lesión en respuesta a la liberación del factor tisular. Este es un cofactor en la activación catalizada del factor X por el factor VIIa, una proteasa de serina que contiene un residuo gla, permite transformación del factor X en factor Xa.
Maritza Gonzalez 88598
ResponderEliminarTrombo vs Coagulo
Los coágulos sanguíneos se presentan cuando la sangre se endurece pasando de líquida a sólida. Un coagulo que se forma dentro de una de las venas o las arterias y corazón se denomina trombo. Un trombo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo. Los trombos arteriales son formados por plaquetas y al microscopio de luz se observan de color blanco, por eso se llaman trombos blancos o arteriales. Los trombos venosos están formados por hilos de fibrina, que atrapan glóbulos rojos y al microscopio se observan de este color.
Fibrinolisis
Es un proceso normal que impide que los coágulos sanguíneos crezcan y causen problemas. La fibrinólisis primaria se refiere a la descomposición normal de los coágulos. La fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa provocando sangrado intenso.
Sistema Fibrinolitico
La fibrinolisis es un proceso enzimático de activadores e inhibidores que regulan la conversión del plasminógeno, en la enzima activa plasmina. La producción de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina.
• Plasminógeno: una glicoproteína que se sintetiza en el hígado y su vida media de 2,2 días. Se une a la fibrina y van a regular la fibrinolisis. También se une a la célula endotelial lo que favorece su activación a nivel local. La activación del plasminógeno a plasmina tiene lugar por acción de los diferentes activadores.
• La precalicreína o el quininógeno de alto peso molecular (HMW-K) pueden inducir la activación del plasminógeno. La activación del factor XII conduce a la generación de calicreina que va a ser un potente activador de la fibrinolisis intrínseca.
• El activador tisular de plasminogeno (t-PA) se fija a la fibrina y forma complejos inactivos entre ambos por tanto, la actividad fibrinolítica plasmática puede aumentar o disminuir dependiendo de una serie de factores.
• Activadores uroquinasas actúan en la activación directa del plasminogeno.
• La activación exógena del plasminogeno se produce por la acción de diferentes sustancias exógenas administradas con fines terapéuticos.
• La Antitrombina VI produce reacciones fibrinolíticas que se desarrollan tras la formación de la fibrina.
Keyla J. Perez Gonzalez 87187
ResponderEliminarACTIVACIÓN PLAQUETARIA
Un enorme número de sustancias puede actuar sobre las plaquetas e inducir su activación [el factor de activación plaquetaria,6 trombina, el tromboxano (TXA2), fosfato de adenosina (ADP),10 epinefrina, vasopresina y serotonina] a través de sus respectivos receptores, pero todas lo hacen a merced de rutas de señalización intracelular que involucran un sistema de proteínas G acoplado a enzimas efectoras comolas fofolipasas A2 (PLA2) y C (PLC) y adenilato y guanilato ciclasas.5, 6, 10
Hasta ahora se han identificado nueve componentes diferentes de la familia de proteínas G en las plaquetas y, dependiendo del tipo de proteína G que participe en la señalización. Se puede activar o inhibir el efecto.6 Un ejemplo lo constituye la regulación por proteína G de la adenilato ciclasa y por tanto de la producción de monofosfato de adenosina cíclico (AMPc). Un incremento de las cantidades de AMPc bloquea la liberación de calcio en el STD e inhibe la función de la plaqueta. Este es el mecanismo que utilizan las prostaciclinas del endotelio vascular sano para inhibir la activación plaquetaria.
Las plaquetas o trombocitos se encuentran en número de 150.000 a 400.000 por mm3 de sangre. Las plaquetas son los elementos formes más pequeños de la sangre. Tienen un diámetro de unas 2μ. Son corpúsculos anucleados con multitud de gránulos citoplasmáticos que son segregados durante su activación. Se forman en la médula ósea, mediante un proceso denominado trombopoyesis. En condiciones normales se forman 40.000 mm3/día.
Las funciones plaquetarias son:
•Mantenimiento de la integridad vascular.
•Interrupción inicial de la hemorragia, mediante la formación del tapón plaquetario, clavo plaquetario o trombo blanco.
•Estabilización del tapón mediante los factores necesarios para la formación de fibrina.
•Retracción del trombo.
•Restauración del endotelio vascular mediante la producción de factores de crecimiento.
Trombopoyesis
•De la célula precursora se diferencian los megacarioblastos, después los megacariocitos y al fragmentarse dan lugar a las plaquetas.
Keyla J Perez Gonzalez 87187
ResponderEliminarHemostasia primaria
Es el conjunto de fenómenos que lleva a la formación del tapón plaquetario, primer paso en la detención de la hemorragia, impidiendo la salida de elementos formes de la sangre. Durante esta fase intervienen dos mecanismos: uno vascular y otro plaquetario.
•a) Espasmo vascular. De manera inmediata a la producción de la rotura del vaso, se produce una potente contracción de las fibras musculares del mismo. El resultado es una vasoconstricción que disminuye el calibre del vaso, e incluso si es pequeño puede llegar a cerrarse, disminuyendo la pérdida de sangre.
•b) Formación del tapón plaquetario. En la formación del tapón plaquetario pueden distinguirse las siguientes etapas:
1.Adhesión o adherencia plaquetaria.
2.Secreción y agregación plaquetaria.
Adhesión o adherencia plaquetaria
Tras la ruptura del endotelio vascular las plaquetas se adhieren a las estructuras subendoteliales, principalmente a las fibras de colágeno que afloran por le superficie rota y entran en contacto con las plaquetas. En este proceso las plaquetas pierden su forma discoide, haciéndose esféricas y emitiendo espículas por medio de las cuales se adhieren al tejido circundante. En el proceso de adhesión se precisan varias glucoproteínas de la membrana plaquetaria, el factor de von Willebrand plasmático y el colágeno y la membrana basal subendoteliales. Este proceso dura muy poco, unos 2-3 segundos.
Secreción y agregación plaquetaria
Se llama agregación al proceso por el cual las plaquetas se fijan unas a otras. Este proceso requiere Ca++ y ADP que deben liberarse de los gránulos plaquetarios mediante un proceso denominado activación o secreción plaquetaria.
Las plaquetas sufren una profunda transformación estructural. Las membranas de los gránulos densos se unen con la membrana plasmática liberando su contenido al exterior y los gránulos α liberan su contenido. Las sustancias liberadas tienen muy diferentes tipos de actividad biológica:
•Estimulan los cambios estrucuturales de las propias plaquetas.
•Aumentan la adherencia plaquetaria y la secreción de más gránulos plaquetarios.
•Aumentan el reclutamiento y activación de más plaquetas.
•Favorecen la agregación y la coagulación.
Esta secreción produce más modificaciones en las plaquetas adheridas y atrae a otras plaquetas, para irse agregando paulatinamente. Las plaquetas se mantienen unidas entre sí por puentes de enlace entre sus membranas y el tejido subendotelial. De esta forma se ha establecido una barrera, aún permeable por los espacios que quedan libres entre las plaquetas, pero que forma una línea de defensa inicial, el tapón plaquetario, o trombo blanco, para la posterior actuación del proceso de la coagulación.
Keyla J Perez Gonzalez 87187
ResponderEliminarHemostasia secundaria o coagulación
Es un proceso que modifica el estado líquido de la sangre dándola una estructura de tipo gel. Consiste en la transformación de una proteína soluble, el fibrinógeno, en una proteía insoluble: la fibrina; formando una malla o red que encierra elementos formes (coágulo), fortaleciendo así la unión entre plaquetas con el objeto de impedir de forma definitiva la hemorragia.
De forma esquemática se puede representar como una cascada enzimática realizada por y sobre proteínas plasmáticas.
Tiene varias fases:
1.Formación de protrombinasa o activador de protrombina.
2.Formación de trombina.
3.Formación de fibrina.
Formación de protrombinasa
Puede seguir dos vías:
•Vía extrínseca, extravascular o exógena (ver esquemas de la presentación en material complementario).
•Vía intrínseca, intravascular o endógena (ver esquemas de la presentación en material complementario).
Las dos vías coinciden activando el factor X para a partir de este punto formar la vía final común. Este factor junto con el factor plaquetario 3, el calcio y el factor V forma un complejo enzimático denominado protrombinasa o activador de la protrombina.
Formación de trombina
Se realiza en una única reacción sobre la protrombina (Factor II).
En la sangre se encuentra presente una proteína inactiva, el Factor I o fibrinógeno.
La trombina cataliza el fraccionamiento de esta molécula formando monómeros de fibrina, solubles e inestables que en presencia de Ca++ y Factor XIII activado se polimerizan; formando un polímero insoluble en forma de red o malla tridimensional que cierra los espacios entre las plaquetas y sella de forma definitiva el tapón plaquetario, dando lugar al trombo rojo o coágulo.
Keyla J Perez Gonzalez 87187
ResponderEliminarFibrinolisis o resolución tras la coagulación
Esta última fase tiene lugar una vez que la pared vascular se ha reconstituido de nuevo, y ya no se requiere la presencia del coágulo. Este proceso se denomina fibrinolisis y consiste en la eliminación de la fibrina. Su importancia es mayor bajo el punto de vista de control en la prevención de la formación de coágulos, que en la eliminación de los mismos. El equilibrio entre la formación de fibrina y su eliminación contribuye a la limitación del proceso hemostático a la región circundante al punto de lesión.
La reacción fundamental es la conversión de una proteína plasmática inactiva el plasminógeno en una activa la plasmina. Esta activación es realizada por factores endógenos como el factor activador del plasminógeno presente en las células endoteliales o la eritrocinasa presente en células sanguíneas.
Sistemas anticoagulantes
La prevención de la coagulación sanguínea en el sistema vascular es un capítulo importante, ya que tan relevante es la formación de un coágulo como su limitación a un tamaño adecuado evitando que se produzca una coagulación indiscriminada.
La superficie endotelial es uno de los mejores factores de seguridad ya que el mantenimiento de su integridad es una garantía para impedir la activación de la hemostasia. Las proteínas de membrana de la célula endotelial repelen los factores de coagulación. Una de estas proteínas es la trombomodulina que actúa como un receptor para la trombina uniéndose a ella y dando lugar a la activación de unas proteínas plasmáticas (C y S) que inactivan factores de coagulación y bloquean la formación de trombina.
Los propios hilos de fibrina absorben entre el 85% y el 90% de la trombina formada, limitando su difusión y su acción proteolítica. Otros inhibidores de la trombina son la antitrombina III que se une a ella inactivándola; y la α2-macroglobulina y la α1-antitripsina. La heparina es un glucosaminoglucano secretado por los mastocitos y leucocitos basófilos que es administrado cuando se requiere una acción anticoagulante rápido. Su mecanismo de acción es unirse a la antitrombina III y potenciar su acción. Otros anticoagulantes funcionan secuestrando el calcio e impidiendo de esta forma la coagulación, como el citrato sódico, oxalato sódico o EDTA sódico. O los denominados anticoagulantes indirectos (cumarinas) que bloquean la absorción de la vitamina K impidiendo la síntesis proteica en el hígado de los factores de coagulación II, VII, IX y X.
Clarilix Rivera Franco 88559Activacion Plaquetaria
ResponderEliminarCuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo. Las células endoteliales producen una proteína llamada factor de von Willebrand (FvW), un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas. Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio. La activación plaquetaria produce el transporte de fosfolípidos cargados a la superficie de la plaqueta, estos fosfolípidos proporcionan una superficie catalítica. El calcio es element esencial para la activación de los factores de coagulación y forma parte de la activacion de mucho de los factores.
Coagulacion de la sangre 1
Luego de una lesion el factor de Von Willebrand se une al colágeno subendotelial. Las plaquetas se van a pegar al factor de Von Willebrand a través de la glicoproteína g1b, solo en el sitio de la lesión. Las plaquetas van a liberar ADP y Calcio necesario para la activación de la cascada de coagulacion. El ADP se pega a su receptor y hace que se exprese en la superficie de la plaquerta la glicoproteína IIb/IIIa. El firbinogeno se une a la glicoproteína IIb/IIIa y crea la agregación plaquetaria.
Coagulacion de la Sangre 2
Comenzando con la cascada intrínseca, después que se forma el trombo primario, este libera ADP que va a activar al Factor XII de coagulación o factor de Hageman. Con la ayuda de calcio el Factor XII va a activar al Factor XI o de Tromboplastina plasmática. Nuevamente calcio entra y El factor XIa produce el Factor IX o de Chrismas. El factor IXa promueve el cambio de el factor X a factor Xa. La cascada extrinsica comienza cuando el factor III tisular promueve la estabilidad del factor VII y en conjunto con el ADP promueven la activación plaquetaria. El factor Va y calcio intervienen para que el Factor Xa de convierta de protrombina a trombina o factor IIa. La trombina es la que cambia el fibrinógeno de la plaqueta por monómeros de fibrina los cuales finalmente promueven la coagulación de las plaquetas y la fibrinosis.
Coagulacion de la sangre 3
Luego de completada la cascada y la activación de la plaquetas, estas plaquetas se agregan entre si para formar un coagulo. Las plaquetas activadas, liberan sustancias químicas que atraen mas plaquetas y esto engrandece la vegetación. A esto se le suma la llegada de macrófagos que forman una capa sobre ellas
Clarilix Rivera Franco 88559
ResponderEliminarCoagulacion de la sangre 4
Comenzando con la cascada intrínseca, después que se forma el trombo primario, este libera ADP que va a activar al Factor XII de coagulación o factor de Hageman. Con la ayuda de calcio el Factor XII va a activar al Factor XI o de Tromboplastina plasmática. Nuevamente calcio entra y El factor XIa produce el Factor IX o de Chrismas. Este factor puede permanecer en circulación hasta 24 horas. Luego el factor IXa promueve el cambio de el factor X a factor Xa. Este factor puede permanecer en circulación hasta 48 horas. La cascada extrinsica comienza cuando el factor III tisular promueve la estabilidad del factor VII y en conjunto con el ADP promueven la activación plaquetaria. El factor Va y calcio intervienen para que el Factor Xa de convierta de protrombina a trombina o factor IIa. La trombina es la que cambia el fibrinógeno de la plaqueta por monómeros de fibrina los cuales finalmente promueven la coagulación de las plaquetas y la fibrinosis
Trombo y Coagulo
Despues de la lesión y una vez realizada la cascada de coagulación, las plaquetas continúan agregándose. Con la llegada de los macrófagos y linfocitos como medio de respuesta inflamatoria, la vegetación queda cubierta capa esponjosa que se conoce como un trombo. Problemas como la hipertensión o cualquier aumento en el flujo de los hematíes a través de la arteria previamente lesionada, puede provocar que este trombo se desprenda, y una vez en circulación se le reconoce como embolo.
Fibrinolisis
La lesión provoca la agregación de plaquetas con el fin de reparar el daño. Esto activa la cascada de coagulación como medio de respuesta inflamatoria importante para la formación de los monómeros de fibrina, que finalmente culminan por sellar la lesión. Esta lesión, aunque reparada se encuentra en mayor riesgo de sufrir rotura y provocarle al paciente un daño mayor, hemorragias, y que entre a circulación el llamado Trombo que pudiera provocar daños mayores en arterias pequeñas distales a la lesión principal.
Sistema Fibrinolitico
El plasminogeno puede ser activado por diferentes enzimas como la calicreina(de la via intriseca), la estreproquina(via exógena), la uroquinasa (de la via endogena) y el activado plasminogeno tisular (via extrinsica). El plasminogeno activado promueve la activación de plamina. La plasmina libera sustancias que promueven la acción de las plaquetas como PDF(antitrombina VI) y el fibrinógeno. Pero este paso suele estar grandemente controlado para evitar la agregación plaquetaria exagerada. Una vez continua aumentado el fibrinógeno y la formación de fibrina, de manera retroalimentación se libera enzimas como la α2 antiplamina y la α2 macroglobulina que controlan la excesiva formación de coagulos.
VIVIANA FLAQUER 85421
ResponderEliminarDIAPOSITIVA #UNO(1)
Activación plaquetaria
Un enorme número de sustancias puede actuar sobre las plaquetas e inducir su activación [el factor de activación plaquetaria,6 trombina, el tromboxano (TXA2), fosfato de adenosina (ADP),10 epinefrina, vasopresina y serotonina] a través de sus respectivos receptores, pero todas lo hacen a merced de rutas de señalización intracelular que involucran un sistema de proteínas G acoplado a enzimas efectoras comolas fofolipasas A2 (PLA2) y C (PLC) y adenilato y guanilato ciclasas.5, 6, 10
Hasta ahora se han identificado nueve componentes diferentes de la familia de proteínas G en las plaquetas y, dependiendo del tipo de proteína G que participe en la señalización. Se puede activar o inhibir el efecto.6 Un ejemplo lo constituye la regulación por proteína G de la adenilato ciclasa y por tanto de la producción de monofosfato de adenosina cíclico (AMPc). Un incremento de las cantidades de AMPc bloquea la liberación de calcio en el STD e inhibe la función de la plaqueta. Este es el mecanismo que utilizan las prostaciclinas del endotelio vascular sano para inhibir la activación plaquetaria.
DIAPOSITIVA #DOS (2)
Coagulación de la Sangre
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse.
Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas.
Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
VIVIANA FLAQUER 85421
ResponderEliminarDIAPOSITIVA # TRES (3)
Coagulación de la sangre
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc.
En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares.
A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
VIVIANA FLAQUER 85421
ResponderEliminarDIAPOSITIVA # CUATRO (4)
El punto en el cual las dos vías se convergen es en la activación del factor X al factor Xa. El factor Xa activa a la protrombina (factor II) para convertirse en trombina (factor IIa). La trombina, a su vez, convierte el fibrinógeno a fibrina. La activación de la trombina ocurre en la superficie de las plaquetas activadas y requiere de la formación de un complejo de protrombinasa. Este complejo está compuesto de los fosfolípidos de las plaquetas: fosfatidilinositol y fosfatidilserina, Ca2+, factores Va y Xa y protrombina. El factor V es un cofactor en la formación del complejo de protrombinasa y tiene un papel similar al del factor VIII en la formación del complejo de tenasa. Como la activación del factor VIII, el factor V es activado al factor Va a través de cantidades diminutas y es inactivado por niveles aumentados de trombina. El factor Va se une a receptores específicos en la superficie de las plaquetas activadas y forma un complejo con la protrombina y el factor Xa.
La protrombina es una proteína de cadena simple de 72,000 Daltons, que contiene diez residuos gla en la región de su N-terminal. Dentro del complejo de protrombinasa, la protrombina es clivada en 2 sitios por el factor Xa. Este clivaje genera una molécula de trombina activa de 2 cadenas que contiene una cadena A y una cadena B las cuales están unidas por una unión simple de disulfuro.
Además de su papel en la activación de la formación del tampón de fibrina, la trombina juega un papel regulador importante en la coagulación. La trombina se une con la trombomodulina presente en la superficie de las células endoteliales formando un complejo que convierte la proteína C a la proteína Ca. La proteína S y la proteína Ca son cofactores que degradan a los factores Va y VIIIa y así limitando la actividad de estos 2 factores en la cascada de la coagulación.
La trombina también se une, resultando en la activación de la señalización, a una clase de receptores acoplados a proteínas G llamados receptores activados por proteasas (siglas en Inglés: PARs), específicamente PAR-1, -3 y -4. Los PARs utilizan un mecanismo único para convertir el producto del clivaje proteolítico extracelular a un evento de señalización intracelular. Los PARs llevan su propio ligando el cual se mantiene inactivo hasta que el clivaje por las proteasas libere al ligando, por ejemplo como es el caso de la trombina. El ligando liberado reacciona con un dominio de unión del ligante del PAR lo que resulta en la activación de varias cascadas de señalización. Estas cascadas de señalización resultan en una liberación aumentada de interleucinas, (ILs), IL-1 e IL-6, una secreción aumentada de la molécula de adhesión intercelular-1 (ICAM-1) y la molécula de adhesión de la célula vascular-1 (VCAM-1). La señalización inducida por la trombina también resulta en un aumento en la activación plaquetaria y en la adhesión de leucocitos.
La trombina también activa al inhibidor de fibrinólisis activado por la trombina (TAFI) y así modulando la fibrinólisis (degradación de los coágulos de fibrina). El TAFI también se conoce como la carboxipeptidasa U (CPU) cuya actividad resulta en la eliminación de las lisinas en la C-terminal de la fibrina parcialmente degradada. Esto conlleva a una falla en la activación del plasminógeno, y así reduciendo la taza de la disolución del coágulo de fibrina (esto se llama fibrinólisis).
VIVIANA FLAQUER 85421
ResponderEliminarDIAPOSITIVA #CINCO (5)
*Vía Intrínseca
Recibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso hemostático.
El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular.
En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación.
En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como la celulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción.
A esta vía es posible subdividirla en tres pasos:
-Formación del factor XIa
En esta etapa participan cuatro proteínas: Precalicreína, Quininógeno de alto peso molecular (HMWK) y los factores XII y XI. Esta etapa no requiere de iones calcio.
Estos cuatro factores se adsorben sobre la superficie cargada negativamente, formando el complejo cebador o de iniciación. De estos factores el XII funciona como verdadero iniciador, ya que si bien es una proenzima, posee una pequeña actividad catalítica que alcanza para activar a la precalicreína convirtiéndola en calicreína.
En segunda instancia la calicreína actúa catalíticamente sobre el factor XII para convertirlo en XIIa, una enzima muchísimo más activa. La actividad catalítica de la calicreína se ve potenciada por el HMWK.
Por último la proteasa XIIa actúa sobre el factor XI para liberar XIa.
-Formación del factor IXa
El factor IX se encuentra en el plasma como una proenzima. En presencia de iones Ca2+ el factor XIa cataliza la ruptura de una unión peptídica en la molécula del factor IX para formar un glucopéptido de 10 KDa y liberar por otro lado al factor IXa.
El factor IX se encuentra ausente en personas con hemofilia tipo B.
-Formación del factor Xa
Sobre la membrana de las plaquetas se forma un complejo constituido por los factores IXa, X y VIII.
Los residuos gamma-carboxiglutamato de los factores IXa y X actúan como quelantes del ion Ca2+, permitiendo que estos componentes formen un complejo unido por medio de puentes de iones calcio y ayudando a que el complejo se ancle a los fosfolípidos de membrana.
Primero se unen los factores X y IXa a la membrana y luego se une el VIII.
El factor VIII es en realidad un homorodímero, formado por cuatro cadenas proteicas, cada una codificada por un gen diferente (VIII:C y VIII:R). El componente VIII:C es conocido como "componente antihemofílico" y actúa como cofactor del IXa en la activación del factor X, el componente VIII:R es el que permite la unión del factor VIII al complejo.
La ausencia del componente antihemofílico causa hemofilia A.
El complejo formado por los factores IXa-X-VIII-Fosfolípidos y Ca2+ actúa sobre el factor X para convertirlo en Xa.
En este punto concluye la vía intrínseca.
VIVIANA FLAQUER 85421
ResponderEliminarCONTINUACION DIAPOSITIVA #CINCO (5)
El factor Xa y la protrombina se adsorben sobre la membrana utilizando iones Ca2+ como puentes. El factor Va se une a la protrombina acelerando la reacción.
El factor Va se produce por la acción de la trombina sobre el factor V en un claro ejemplo de una reacción que va acelerándose a medida que progresa (reacción autoacelerada).
-Formación de fibrina
El fibrinógeno (factor I) es una glicoproteína compuesta por seis cadenas polipeptídicas: dos A-alfa, dos B-beta y dos gamma; unidas entre sí por puentes disulfuro.
Se trata de una molécula alargada y simétrica formada por tres dominios globulares conectados por segmentos fibrilares.
Cada mitad de la molécula se encuentra formada por tres cadenas (A-alfa, B-beta y gamma) que se enrollan en una triple hélice muy compacta en los sectores fibrilares. Los extremos amino de las seis cadenas se reúnen en el dominio globular central.
En un hecho que parecería muy curioso, los extremos N-terminales de las cadenas A-alfa y B-beta emergen como cabos libres del dominio globular central.
Representación de la molécula de fibrinógeno y cómo, al eliminarse los fibrinopéptidos, polimeriza para formar un agregado de fibrina.
Estas cadenas son muy ricas en aspartato y glutamato, además las cadenas B-beta poseeen en esta región residuos tirosina-O-sulfato formados postraduccionalmente. Estos residuos con una alta tendencia a adquirir carga negativa contribuyen a formar una región central con una muy alta densidad de carga.
Esta región electronegativa central es la responsable de la repulsión entre moléculas de fibrina que las mantiene en solución.
La trombina ataca los enlaces arginina-glicina presentes en estos "cabos libres", separando cuatro péptidos; dos segmentos A de 18 aminoácidos cada uno (provenientes de las cadenas A-alfa), y dos segmentos B de 20 aminoácidos (provenientes de las cadenas B-beta). A estos péptidos se los suele denominar "fibrinopéptidos".
El resto que queda de la molécula es un monómero de fibrina de composición alfa2beta2gamma2.
Al eliminarse los fibinopéptidos desaparecen las fuerzas de repulsión intermoleculares con lo que los monómeros de fibrina tienden a agruparse espontáneamente formando asociaciones altamente ordenadas.
Los monómeros se disponen uno a continuación del otro, cabeza con cabeza en forma de largas hebras. Estas hebras a su vez forman manojos, emparejándose con otras hebras de tal manera que la región central de los monómeros de fibrina de una se encuentra rodeada por las cabezas de los monómeros de fibrina de las otras.
Este emparejamiento se hace posible gracias a interaciones de tipo electrostático y puente hidrógeno entre las regiones centrales de los monómeros de una y las cabezas globulares de otras.
-Entrecruzamiento de la fibrina
Los haces paralelos de fibrina polimerizada forman una asociación laxa, que se encuentra en equilibrio con la forma monomérica de la molécula; por lo que sería imposible que cumplieran su papel de formar un coágulo estable sin reforzar esta estructura por medio de enlaces covalentes entre hebras vecinas.
La formación de estos "puentes" covalentes intercatenarios es catalizada por la enzima transglutaminasa (conocida también como factor XIIIa).
La transglutaminidasa cataliza la formación de enlaces amida entre restos glutamina y lisina de hebras próximas entre sí. En la reacción se libera amoníaco en forma de ion amonio (NH4+).
Esta enzima se forma a partir del factor XIII por acción de la trombina.
VIVIANA FLAQUER 85421
ResponderEliminarDIAPOSITIVA #SEIS (6)
Trombo & Cuagulo
Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida.
Un coágulo sanguíneo que se forma dentro de una de las venas o las arterias se denomina trombo y también se puede formar en el corazón.
Un trombo que se desprende y viaja desde un lugar en el cuerpo a otro se llama émbolo.Un trombo o émbolo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo.
Una obstrucción en una arteria puede impedir que el oxígeno llegue a los tejidos en esa área, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede provocar daños en los tejidos o la muerte.
Una obstrucción en una vena generalmente provocará acumulación de líquido e hinchazón.
Las situaciones en las que es más probable que se forme un coágulo de sangre abarcan:
Estar en reposo en cama por largo tiempo.
Cruzar las piernas durante largos periodos al estar sentado o sentarse durante mucho tiempo, como en un avión o en un vehículo.
Durante y después del embarazo.
No tener suficiente agua en el cuerpo (deshidratación).
Tomar píldoras anticonceptivas u hormonas estrógeno (especialmente en las mujeres que fuman).
Utilizar un catéter intravenoso por largo tiempo.
Los coágulos de sangre también son más probables en personas con cáncer, cirugía o lesión recientes, obesidad y enfermedades del hígado o del riñón.
Una acumulación de colesterol que estreche una arteria puede cambiar o reducir el flujo de sangre, facilitando la formación de un coágulo sanguíneo o de un trombo.
Las afecciones que se transmiten de padres a hijos (hereditarias) pueden hacerlo más propenso a la formación de coágulos sanguíneos anormales. Los trastornos hereditarios que afectan la coagulación son:
Trombofilia por factor V de Leiden
Mutación de la protrombina G20210A
Otras afecciones raras como deficiencias de antitrombina III, proteína C y S
Un coágulo de sangre puede bloquear una arteria o una vena en el corazón, afectando:
El corazón (angina de pecho o un ataque cardíaco)
Los intestinos (isquemia mesentérica) o trombosis venosa mesentérica
Los riñones (trombosis de la vena renal)
Las arterias de las piernas o brazos
Las piernas (trombosis venosa profunda)
Los pulmones (embolia pulmonar)
El cuello o el cerebro (accidente cerebrovascular)
Los trombos y émbolos pueden fijarse firmemente a un vaso sanguíneo y pueden bloquear parcial o completamente el flujo de sangre a dicho vaso.
Un bloqueo en el vaso sanguíneo impide que el flujo normal de sangre y oxígeno llegue a los tejidos en ese sitio, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede ocasionar daño tisular o necrosis en esa área.
VIVIANA FLAQUER 85421
ResponderEliminarDIAPOSITIVA #SIETE (7)
Fibrinólisis
Después de que el coágulo se ha establecido, comienza la reparación de los tejidos afectados con el proceso de cicatrización. Para hacer posible esto el coágulo es colonizado por células que formarán nuevos tejidos y en el proceso va siendo degradado.
La degradación de la fibrina (fibrinólisis), componente mayoritaria del coágulo, es catalizada por la enzima plasmina, una serina proteasa que ataca las uniones peptídicas en la región triple hélice de los monómeros de fibrina.
La plasmina se genera a partir del plasminógeno, un precursor inactivo; activándose tanto por la acción de factores intrínsecos (propios de la cascada de coagulación) como extrínsecos, el más importante de los cuales es producido por el endotelio vascular. Se le denomina "activador tisular del plasminógeno" (t-PA).
El gen de este factor ha sido clonado y actualmente se puede obtener la proteína producida por tecnología de ADN recombinante.
Este factor suele utilizarse en clínica para favorecer la disolución de trombos.
DIAPOSITIVA #OCHO (8)
Sistema Fibrinolitico
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos.
La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón.
La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca:
En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA.
En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno.
La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Grace Marie Fernandez 83012
ResponderEliminarLas plaquetas son células producidas por los megacariocitos en la médula ósea mediante el proceso de fragmentación citoplasmática, circulan por la sangre y tiene un papel muy importante en la coagulación. Para ello forman nudos en la red de fibrina, liberan substancias importantes para acelerar la coagulación y aumentan la retracción del coágulo sanguíneo. En las heridas las plaquetas aceleran la coagulación, y además al aglutinarse obstruyen pequeños vasos, y engendran substancias que los contraen.
Procedimiento de obtención: Para realizar este análisis no se precisa estar en ayunas.
Se puede realizar la toma en un lugar apropiado (consulta, clínica, hospital) pero en ocasiones se realiza en el propio domicilio del paciente. Para realizar la toma se precisa de localizar una vena apropiada y en general se utilizan las venas situadas en la flexura del codo. La persona encargada de tomar la muestra utilizará guantes sanitarios, una aguja (con una jeringa o tubo de extracción).
Le pondrá un tortor (cinta de goma-látex) en el brazo para que las venas retengan más sangre y aparezcan más visibles y accesibles. Limpiará la zona del pinchazo con un antiséptico y mediante una palpación localizará la vena apropiada y accederá a ella con la aguja. Le soltarán el tortor.
Cuando la sangre fluya por la aguja el sanitario realizará una aspiración (mediante la jeringa o mediante la aplicación de un tubo con vacío). Si se requiere varias muestras para diferentes tipos de análisis se le extraerá más o menos sangre o se aplicarán diferentes tubos de vacío.
Al terminar la toma, se extrae la aguja y se presiona la zona con una torunda de algodón o similar para favorecer la coagulación y se le indicará que flexione el brazo y mantenga la zona presionada con un esparadrapo durante unas horas.
Problemas y posibles riesgos:
La obtención mediante un pinchazo de la vena puede producir cierto dolor.
La posible dificultad en encontrar la vena apropiada puede dar lugar a varios pinchazos.
Aparición de un hematoma (moratón o cardenal) en la zona de extracción, suele deberse a que la vena no se ha cerrado bien tras la presión posterior y ha seguido saliendo sangre produciendo este problema. Puede aplicarse una pomada tipo Hirudoid® o Trombocid® en la zona.
Inflamación de la vena (flebitis), a veces la vena se ve alterada, bien sea por una causa meramente física o por que se ha infectado. Se deberá mantener la zona relajada unos días y se puede aplicar una pomada tipo Hirudoid® o Trombocid® en la zona. Si el problema persiste o aparece fiebre deberá consultarlo con su médico.
Valores normales: Las alteraciones en el número de plaquetas así como en su tamaño pueden ser clave del diagnóstico. Hay una gran variación en el rango normal del recuento de plaquetas.
Valores normales: De 150.000 a 400.000/mm3
Significado de los resultados anormales: La disminución en el número de plaquetas (por debajo del límite menor normal) se denomina trombocitopenia y el aumento en el número de las mismas (superior al límite normal más alto) se llama trombocitosis. Cuando existe una trombocitopenia aislada, la causa más común es la destrucción inmune, pero existen trombocitopenias asociadas a un gran número de otras enfermedades como son:
Coagulación intravascular diseminada (C.IV.D)
Anemia hemolítica microangiopática
Hiperesplenismo (exceso de función del bazo)
Disminución de la producción en el caso de anemia aplástica,
Invasión de la médula ósea por enfermedades malignas como leucemias, neuroblastoma, linfoma.
Quimioterapia por cáncer
Púrpura trombocitopénica idiopática (PTI)
Leucemia
Prótesis de válvula coronaria
Transfusión de sangre
Choque anafiláctico
Algunas infecciones que producen hemorragias (púrpuras con trombocitopenia), en las que se hallan muy disminuidas.
Grace Marie Fernandez 83012:
ResponderEliminarLa trombocitosis es el aumento en el recuento de plaquetas y puede ser secundario Las infecciones suelen ser la causa más frecuente (virales, bacterianas o por micoplasma), pero existen muchas otras enfermedades que se asocian a trombocitosis como son:
Anemia por déficit de hierro
Enfermedad de Kawasaki
Síndrome nefrótico
Síndrome post-esplenectomía (tras extraer el bazo)
Traumatismos
Tumores
Trombocitosis primaria
La coagulación es el proceso por el cual se forma un coágulo sanguíneo. Comienza en respuesta a una lesión en un vaso sanguíneo. En el proceso de coagulación se producen una serie de reacciones en cadena en las que participan varios tipos celulares y proteínas solubles de la sangre con el objetivo de formar un coágulo para evitar la pérdida excesiva de sangre. Un coágulo consiste en una red de proteínas insolubles como la fibrina con plaquetas y células atrapadas que bloquea la salida de sangre hasta que se repare el tejido. Se distinguen dos rutas de activación de la cascada de coagulación conocidas como vía extrínseca y vía intrínseca. Hacen referencia al lugar donde se inicia la cascada de coagulación: el interior de un vaso sanguíneo (intrínseca) o fuera de un vaso sanguíneo (extrínseca). Ambas vías convergen en la activación del factor Xa que transforma la protrombina en trombina. En el paso siguiente la trombina genera fibrina a partir de fibrinógeno.
Grace Marie Fernandez 83012:
ResponderEliminarEn el proceso de coagulación participan los siguientes elementos:
Células que presentan el factor tisular (TF): Estas células no están normalmente en contacto con elementos solubles de la sangre. Cuando se produce una lesión con pérdida de sangre, el reconocimiento entre el TF y las proteínas solubles dispara la cascada de la coagulación.
Plaquetas: Son fragmentos celulares derivados del megacariocito. Participan en el proceso de coagulación en las fases de amplificación y propagación y forman parte del coágulo. En sus membranas se dan muchas de las reacciones del proceso de coagulación y secretan varios elementos imprescindibles durante el proceso.
Factores de coagulación: Son un conjunto de proteínas que participan en las reacciones enzimáticas encadenadas que hacen posible la cascada de la coagulación. Las proteínas de la cascada se van activando por proteolisis adquiriendo actividad proteasa y activando por proteolisis a la siguiente proteína de la cascada de coagulación. La activación de esta cascada lleva a la producción de grandes cantidades de trombina que posibilitan la formación del coágulo. Algunos de estos factores se asocian entre sí en la membrana de plaquetas y de otros tipos celulares que participan en el proceso de coagulación
Sistema anticoagulante: Para evitar la formación de trombos (coágulos fijos dentro de los vasos) o émbolos (coágulos móviles que viajan por el torrente sanguíneo) existen una serie de proteínas que confinan la formación del coágulo a la zona lesionada. Algunos elementos de estos sistemas son la proteína C, la proteína S, la antitrombina (AT) y la trombomodulina (TM).
El proceso de coagulación se puede esquematizar en las siguientes fases:
Fase de iniciación. En esta fase es fundamental la formación de pequeñas cantidades de trombina. La lesión de un vaso sanguíneo expone las proteínas solubles de la sangre a las células de los tejidos circundantes y a la matriz extracelular. Estos contactos van a generar una serie de reacciones que constituyen la fase de iniciación. La cascada de reacciones que tiene lugar en esta fase comienza cuando el factor VIIa (factor VII activo) se une a los factores tisulares (TF) en las membranas de las células que están fuera de los vasos originando el complejo VIIa/TF. Este complejo activa a su vez pequeñas cantidades de factor IX y X. El factor Xa (factor X activo) se asocia con el factor Va (factor V activo) en la superficie de las células que presentan los factores tisulares. Se forma un complejo protrombinasa que origina pequeñas cantidades de trombina (factor IIa). El factor V puede ser activado por el propio factor Xa, por ciertas proteasas de la matriz extracelular o puede ser liberado directamente por parte de las plaquetas al contactar con el colágeno u otros componentes de la matriz extracelular.
Grace Marie Fernandez 83012:
ResponderEliminarNormalmente el factor Xa permanece unido a la membrana de las células que presentan factores tisulares para protegerse de la degradación. En caso de producirse su disociación y pasar al torrente sanguíneo es rápidamente inhibido en la fase fluida por inhibidores de la ruta de los factores tisulares o por antitrombina evitando que sea activo fuera de la zona de la lesión. De esta forma, la presencia de inhibidores focaliza y limita eficientemente la actividad del factor Xa únicamente a las superficies celulares donde se produce la lesión. El factor IXa puede moverse desde la célula en que se origina a través de la fase fluida hacia una plaqueta vecina u otra superficie celular ya que es inhibido mucho más lentamente por antitrombina.
Fase de amplificación. En esta fase se produce la activación de las plaquetas gracias a la pequeña cantidad de trombina generada en la fase de iniciación. En esta fase factores en la superficie de las plaquetas provocan la producción masiva de trombina. La fase de amplificación tiene lugar sólo si la lesión permite la salida de plaquetas y del complejo VIII/vWF (factor von Willebrand). Durante la activación de las plaquetas se secretan formas parcialmente activadas de factor V en la superficie. En esta fase también se disocia el complejo VIII/vWF, permitiendo al factor vWF mediar la adhesión y agregación adicional de plaquetas en el sitio de la lesión y al factor VIII activarse y unirse a la membrana de las plaquetas. La trombina generada también activa el factor XI en la superficie de la plaqueta durante esta fase.
Fase de propagación. En esta fase se produce trombina de forma masiva para la formación de un coágulo estable. También acuden gran número de plaquetas al lugar de la lesión. En esta fase sólo participan las plaquetas activadas. El factor IXa, generado en la fase de iniciación, se une al factor VIIIa en la superficie de las plaquetas. El factor XIa en la superficie de plaquetas permite la unión de más cantidad de factor IXa. El factor Xa generado por el complejo IXa/VIIIa formado en la membrana de las plaquetas se asocia rápidamente con el factor Va expresado por las plaquetas en la fase de amplificación. Al ensamblarse la protrombinasa (Xa/Va) provoca una masiva producción de trombina en cantidad suficiente para que se forme el coágulo.
Grace Marie Fernandez 83012:
ResponderEliminarUna vez formado el coágulo de fibrina y plaquetas en la zona lesionada, el proceso ha de ser controlado para evitar la oclusión trombótica en las zonas no dañadas. La proteína C, la proteína S y la trombomodulina constituyen un importante sistema de control que restringe la coagulación a la zona de la lesión. Durante el proceso de coagulación parte de la trombina formada puede difundir por los vasos sanguíneos. Cuando llega a una célula endotelial intacta se une a la trombomodulina en la superficie endotelial. El complejo trombomodulina/trombina activa a la proteína C que se une a la proteína S e inactiva los factores Va y VIIIa. De esta forma se detiene la generación de trombina en las zonas intactas. La proteína C y la proteína S son mucho más eficientes inactivando al factor Va en la superficie de las células endoteliales que en la de las plaquetas. Otro sistema para prevenir la generación de trombina en células endoteliales de zonas no dañadas es la acción de los inhibidores de proteasa antitrombina e inhibidores de la ruta de los factores tisulares (TFPI: Tissue Factor Pathway Inhibitor) que ayudan a confinar la formación de trombina a las áreas alrededor de la lesión. Los inhibidores de proteasa AT-III y TFPI pueden inhibir rápidamente proteasas generadas cerca de un endotelio intacto.
Existen diversas enfermedades derivadas de fallos en algunos de estos elementos que participan en el proceso de coagulación. Pueden tener un origen genético o no y ser o no reversibles. Existen tres grupos de genes asociados a defectos en la coagulación:
Genes responsables de la adhesión, activación y agregación plaquetaria.
Genes de biosíntesis de factores y cofactores de coagulación sanguínea.
Genes involucrados en el sistema de anticoagulación y fibrinolisis.
La hemofilia es un ejemplo de enfermedad causada por un defecto genético en un componente del proceso de coagulación (factor VIII en la hemofilia A y factor IX en la hemofilia B).
Los fármacos anticoagulantes que se han empleado tradicionalmente tienen sus limitaciones. Actualmente se están desarrollando nuevos anticoagulantes inhibidores del factor Xa e inhibidores directos de la trombina. Varios de ellos aún están en fase de ensayo clínico para prevención y tratamiento de la trombosis: "Dabigatran Etexilate" es un fármaco oral que se convierte en "dabigatran", un inhibidor de la trombina. La eficacia y seguridad de esta sustancia ha sido evaluada en la fase II del ensayo clínico y se encuentra en la fase III de prevención y tratamiento de la enfermedad tromboembólica venosa (VTE: Venous Thromboembolism Events) y prevención de apoplejías derivadas de fibrilación auricular. Otra diana terapéutica para el diseño de drogas anticoagulantes es el factor Xa. "Fondaparinux", es un pentasacárido obtenido por síntesis química que se une de forma rápida y específica a antitrombina en la sangre, induciendo un cambio conformacional en la antitrombina. Este cambio conformacional incrementa su afinidad por el factor Xa potenciando su función inhibidora unas 300 veces. Además cada molécula de fármaco puede modificar a varias moléculas de antitrombina. Esta sustancia ha sido probada en numerosos ensayos clínicos de fase III con diferentes dosis, obteniéndose una reducción de un 50% en la incidencia de enfermedad tromboembólica venosa.
Grace Marie Fernandez 83012:
ResponderEliminarLa fibrinólisis primaria se refiere a la descomposición normal de los coágulos, mientras que la fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa.
Cuando se produce demasiado rápidamente, puede provocar hemorragias espectaculares (fibrinólisis hemorrágica) intra o postoperatorias (particularmente en Cirugía Torácica), o después de un parto, de un aborto o de un choque traumático; o bien puede provocar hemorragias menos importantes (hematuria) en el curso de ciertos cánceres (próstata, páncreas, estómago), de las cirrosis y de las leucosis. Obedece a la liberación de activadores de la profibrinolisina, o bien a la producción de un fermento proteolítico por los tejidos enfermos.
Puede ser primitiva aunque por lo general es secundaria, reactiva, en el curso de un síndrome de coagulación intravascular diseminada.
Los coágulos de sangre se forman a partir de una proteína llamada fibrina. La descomposición de la fibrina (fibrinólisis) puede incrementarse bajo ciertas condiciones, tales como:
Infecciones bacterianas
Ejercicio intenso
Glucemia baja
Oxigenación insuficiente a los tejidos
ENMANUEL REYES S. #87419
ResponderEliminarDIAPOSITIVA #1
ACTIVACIÓN PLAQUETARIA.
Un enorme número de sustancias puede actuar sobre las plaquetas e inducir su activación [el factor de activación plaquetaria,6 trombina, el tromboxano (TXA2), fosfato de adenosina (ADP),10 epinefrina, vasopresina y serotonina] a través de sus respectivos receptores, pero todas lo hacen a merced de rutas de señalización intracelular que involucran un sistema de proteínas G acoplado a enzimas efectoras comolas fofolipasas A2 (PLA2) y C (PLC) y adenilato y guanilato ciclasas.5, 6, 10
Hasta ahora se han identificado nueve componentes diferentes de la familia de proteínas G en las plaquetas y, dependiendo del tipo de proteína G que participe en la señalización. Se puede activar o inhibir el efecto.6 Un ejemplo lo constituye la regulación por proteína G de la adenilato ciclasa y por tanto de la producción de monofosfato de adenosina cíclico (AMPc). Un incremento de las cantidades de AMPc bloquea la liberación de calcio en el STD e inhibe la función de la plaqueta. Este es el mecanismo que utilizan las prostaciclinas del endotelio vascular sano para inhibir la activación plaquetaria.
DIAPOSITIVA #2
COAGULACIÓN DE LA SANGRE.
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse.
Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas.
Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
ENMANUEL REYES S. #87419
ResponderEliminarDIAPOSITIVA # 3
COAGULACIÓN DE LA SANGRE
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc.
En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares.
A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
ENMANUEL REYES S. #87419
ResponderEliminarDIAPOSITIVA # 4
El punto en el cual las dos vías se convergen es en la activación del factor X al factor Xa. El factor Xa activa a la protrombina (factor II) para convertirse en trombina (factor IIa). La trombina, a su vez, convierte el fibrinógeno a fibrina. La activación de la trombina ocurre en la superficie de las plaquetas activadas y requiere de la formación de un complejo de protrombinasa. Este complejo está compuesto de los fosfolípidos de las plaquetas: fosfatidilinositol y fosfatidilserina, Ca2+, factores Va y Xa y protrombina. El factor V es un cofactor en la formación del complejo de protrombinasa y tiene un papel similar al del factor VIII en la formación del complejo de tenasa. Como la activación del factor VIII, el factor V es activado al factor Va a través de cantidades diminutas y es inactivado por niveles aumentados de trombina. El factor Va se une a receptores específicos en la superficie de las plaquetas activadas y forma un complejo con la protrombina y el factor Xa.
La protrombina es una proteína de cadena simple de 72,000 Daltons, que contiene diez residuos gla en la región de su N-terminal. Dentro del complejo de protrombinasa, la protrombina es clivada en 2 sitios por el factor Xa. Este clivaje genera una molécula de trombina activa de 2 cadenas que contiene una cadena A y una cadena B las cuales están unidas por una unión simple de disulfuro.
Además de su papel en la activación de la formación del tampón de fibrina, la trombina juega un papel regulador importante en la coagulación. La trombina se une con la trombomodulina presente en la superficie de las células endoteliales formando un complejo que convierte la proteína C a la proteína Ca. La proteína S y la proteína Ca son cofactores que degradan a los factores Va y VIIIa y así limitando la actividad de estos 2 factores en la cascada de la coagulación.
La trombina también se une, resultando en la activación de la señalización, a una clase de receptores acoplados a proteínas G llamados receptores activados por proteasas (siglas en Inglés: PARs), específicamente PAR-1, -3 y -4. Los PARs utilizan un mecanismo único para convertir el producto del clivaje proteolítico extracelular a un evento de señalización intracelular. Los PARs llevan su propio ligando el cual se mantiene inactivo hasta que el clivaje por las proteasas libere al ligando, por ejemplo como es el caso de la trombina. El ligando liberado reacciona con un dominio de unión del ligante del PAR lo que resulta en la activación de varias cascadas de señalización. Estas cascadas de señalización resultan en una liberación aumentada de interleucinas, (ILs), IL-1 e IL-6, una secreción aumentada de la molécula de adhesión intercelular-1 (ICAM-1) y la molécula de adhesión de la célula vascular-1 (VCAM-1). La señalización inducida por la trombina también resulta en un aumento en la activación plaquetaria y en la adhesión de leucocitos.
La trombina también activa al inhibidor de fibrinólisis activado por la trombina (TAFI) y así modulando la fibrinólisis (degradación de los coágulos de fibrina). El TAFI también se conoce como la carboxipeptidasa U (CPU) cuya actividad resulta en la eliminación de las lisinas en la C-terminal de la fibrina parcialmente degradada. Esto conlleva a una falla en la activación del plasminógeno, y así reduciendo la taza de la disolución del coágulo de fibrina (esto se llama fibrinólisis).
ENMANUEL REYES S. #87419
ResponderEliminarDIAPOSITIVA #5
*Vía Intrínseca
Recibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso hemostático.
El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular.
En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación.
En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como la celulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción.
A esta vía es posible subdividirla en tres pasos:
-Formación del factor XIa
En esta etapa participan cuatro proteínas: Precalicreína, Quininógeno de alto peso molecular (HMWK) y los factores XII y XI. Esta etapa no requiere de iones calcio.
Estos cuatro factores se adsorben sobre la superficie cargada negativamente, formando el complejo cebador o de iniciación. De estos factores el XII funciona como verdadero iniciador, ya que si bien es una proenzima, posee una pequeña actividad catalítica que alcanza para activar a la precalicreína convirtiéndola en calicreína.
En segunda instancia la calicreína actúa catalíticamente sobre el factor XII para convertirlo en XIIa, una enzima muchísimo más activa. La actividad catalítica de la calicreína se ve potenciada por el HMWK.
Por último la proteasa XIIa actúa sobre el factor XI para liberar XIa.
-Formación del factor IXa
El factor IX se encuentra en el plasma como una proenzima. En presencia de iones Ca2+ el factor XIa cataliza la ruptura de una unión peptídica en la molécula del factor IX para formar un glucopéptido de 10 KDa y liberar por otro lado al factor IXa.
El factor IX se encuentra ausente en personas con hemofilia tipo B.
-Formación del factor Xa
Sobre la membrana de las plaquetas se forma un complejo constituido por los factores IXa, X y VIII.
Los residuos gamma-carboxiglutamato de los factores IXa y X actúan como quelantes del ion Ca2+, permitiendo que estos componentes formen un complejo unido por medio de puentes de iones calcio y ayudando a que el complejo se ancle a los fosfolípidos de membrana.
Primero se unen los factores X y IXa a la membrana y luego se une el VIII.
El factor VIII es en realidad un homorodímero, formado por cuatro cadenas proteicas, cada una codificada por un gen diferente (VIII:C y VIII:R). El componente VIII:C es conocido como "componente antihemofílico" y actúa como cofactor del IXa en la activación del factor X, el componente VIII:R es el que permite la unión del factor VIII al complejo.
La ausencia del componente antihemofílico causa hemofilia A.
El complejo formado por los factores IXa-X-VIII-Fosfolípidos y Ca2+ actúa sobre el factor X para convertirlo en Xa.
En este punto concluye la vía intrínseca.
CONTINUACION DE LA DIAPOSITIVA #5
ResponderEliminarEl factor Xa y la protrombina se adsorben sobre la membrana utilizando iones Ca2+ como puentes. El factor Va se une a la protrombina acelerando la reacción.
El factor Va se produce por la acción de la trombina sobre el factor V en un claro ejemplo de una reacción que va acelerándose a medida que progresa (reacción autoacelerada).
-Formación de fibrina
El fibrinógeno (factor I) es una glicoproteína compuesta por seis cadenas polipeptídicas: dos A-alfa, dos B-beta y dos gamma; unidas entre sí por puentes disulfuro.
Se trata de una molécula alargada y simétrica formada por tres dominios globulares conectados por segmentos fibrilares.
Cada mitad de la molécula se encuentra formada por tres cadenas (A-alfa, B-beta y gamma) que se enrollan en una triple hélice muy compacta en los sectores fibrilares. Los extremos amino de las seis cadenas se reúnen en el dominio globular central.
En un hecho que parecería muy curioso, los extremos N-terminales de las cadenas A-alfa y B-beta emergen como cabos libres del dominio globular central.
Representación de la molécula de fibrinógeno y cómo, al eliminarse los fibrinopéptidos, polimeriza para formar un agregado de fibrina.
Estas cadenas son muy ricas en aspartato y glutamato, además las cadenas B-beta poseeen en esta región residuos tirosina-O-sulfato formados postraduccionalmente. Estos residuos con una alta tendencia a adquirir carga negativa contribuyen a formar una región central con una muy alta densidad de carga.
Esta región electronegativa central es la responsable de la repulsión entre moléculas de fibrina que las mantiene en solución.
La trombina ataca los enlaces arginina-glicina presentes en estos "cabos libres", separando cuatro péptidos; dos segmentos A de 18 aminoácidos cada uno (provenientes de las cadenas A-alfa), y dos segmentos B de 20 aminoácidos (provenientes de las cadenas B-beta). A estos péptidos se los suele denominar "fibrinopéptidos".
El resto que queda de la molécula es un monómero de fibrina de composición alfa2beta2gamma2.
Al eliminarse los fibinopéptidos desaparecen las fuerzas de repulsión intermoleculares con lo que los monómeros de fibrina tienden a agruparse espontáneamente formando asociaciones altamente ordenadas.
Los monómeros se disponen uno a continuación del otro, cabeza con cabeza en forma de largas hebras. Estas hebras a su vez forman manojos, emparejándose con otras hebras de tal manera que la región central de los monómeros de fibrina de una se encuentra rodeada por las cabezas de los monómeros de fibrina de las otras.
Este emparejamiento se hace posible gracias a interaciones de tipo electrostático y puente hidrógeno entre las regiones centrales de los monómeros de una y las cabezas globulares de otras.
-Entrecruzamiento de la fibrina
Los haces paralelos de fibrina polimerizada forman una asociación laxa, que se encuentra en equilibrio con la forma monomérica de la molécula; por lo que sería imposible que cumplieran su papel de formar un coágulo estable sin reforzar esta estructura por medio de enlaces covalentes entre hebras vecinas.
La formación de estos "puentes" covalentes intercatenarios es catalizada por la enzima transglutaminasa (conocida también como factor XIIIa).
La transglutaminidasa cataliza la formación de enlaces amida entre restos glutamina y lisina de hebras próximas entre sí. En la reacción se libera amoníaco en forma de ion amonio (NH4+).
Esta enzima se forma a partir del factor XIII por acción de la trombina.
ENMANUEL REYES S. #87419
ResponderEliminarDIAPOSITIVA #6
TROMBO & CUAGULO
Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida.
Un coágulo sanguíneo que se forma dentro de una de las venas o las arterias se denomina trombo y también se puede formar en el corazón.
Un trombo que se desprende y viaja desde un lugar en el cuerpo a otro se llama émbolo.Un trombo o émbolo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo.
Una obstrucción en una arteria puede impedir que el oxígeno llegue a los tejidos en esa área, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede provocar daños en los tejidos o la muerte.
Una obstrucción en una vena generalmente provocará acumulación de líquido e hinchazón.
Las situaciones en las que es más probable que se forme un coágulo de sangre abarcan:
Estar en reposo en cama por largo tiempo.
Cruzar las piernas durante largos periodos al estar sentado o sentarse durante mucho tiempo, como en un avión o en un vehículo.
Durante y después del embarazo.
No tener suficiente agua en el cuerpo (deshidratación).
Tomar píldoras anticonceptivas u hormonas estrógeno (especialmente en las mujeres que fuman).
Utilizar un catéter intravenoso por largo tiempo.
Los coágulos de sangre también son más probables en personas con cáncer, cirugía o lesión recientes, obesidad y enfermedades del hígado o del riñón.
Una acumulación de colesterol que estreche una arteria puede cambiar o reducir el flujo de sangre, facilitando la formación de un coágulo sanguíneo o de un trombo.
Las afecciones que se transmiten de padres a hijos (hereditarias) pueden hacerlo más propenso a la formación de coágulos sanguíneos anormales. Los trastornos hereditarios que afectan la coagulación son:
Trombofilia por factor V de Leiden
Mutación de la protrombina G20210A
Otras afecciones raras como deficiencias de antitrombina III, proteína C y S
Un coágulo de sangre puede bloquear una arteria o una vena en el corazón, afectando:
El corazón (angina de pecho o un ataque cardíaco)
Los intestinos (isquemia mesentérica) o trombosis venosa mesentérica
Los riñones (trombosis de la vena renal)
Las arterias de las piernas o brazos
Las piernas (trombosis venosa profunda)
Los pulmones (embolia pulmonar)
El cuello o el cerebro (accidente cerebrovascular)
Los trombos y émbolos pueden fijarse firmemente a un vaso sanguíneo y pueden bloquear parcial o completamente el flujo de sangre a dicho vaso.
Un bloqueo en el vaso sanguíneo impide que el flujo normal de sangre y oxígeno llegue a los tejidos en ese sitio, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede ocasionar daño tisular o necrosis en esa área.
DIAPOSITIVA #7
FIBRINÓLISIS
Después de que el coágulo se ha establecido, comienza la reparación de los tejidos afectados con el proceso de cicatrización. Para hacer posible esto el coágulo es colonizado por células que formarán nuevos tejidos y en el proceso va siendo degradado.
La degradación de la fibrina (fibrinólisis), componente mayoritaria del coágulo, es catalizada por la enzima plasmina, una serina proteasa que ataca las uniones peptídicas en la región triple hélice de los monómeros de fibrina.
La plasmina se genera a partir del plasminógeno, un precursor inactivo; activándose tanto por la acción de factores intrínsecos (propios de la cascada de coagulación) como extrínsecos, el más importante de los cuales es producido por el endotelio vascular. Se le denomina "activador tisular del plasminógeno" (t-PA).
El gen de este factor ha sido clonado y actualmente se puede obtener la proteína producida por tecnología de ADN recombinante.
Este factor suele utilizarse en clínica para favorecer la disolución de trombos.
ENMANUEL REYES S. #87419
ResponderEliminarDIAPOSITIVA #8
SISTEMA FIBRINOLITICO
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos.
La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón.
La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca:
En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA.
En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno.
La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarPlaquetas, Coagulación y Fibrinolisis
PLAQUETA
Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro, derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos.
Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva.
Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebro vascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores; cualquier anormalidad o enfermedad de las plaquetas es denominada trombocitopatía, la cual puede ser, ya sea un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Hay desórdenes que pueden reducir el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos, pero en cambio otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragia.
Las plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular; Estos dos factores de crecimiento han demostrado jugar un papel significativo en la regeneración y reparación del tejido conectivo; Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.
DESCUBRIMIENTO
Max Schultze (1825-1874), un anatomista alemán, marcó la historia del descubrimiento de las plaquetas. Sin embargo los glóbulos rojos, o eritrocitos, ya eran conocidos desde van Leeuwenhoek, Schultze fue primero en publicar una descripción de las plaquetas.11Él describió "esférulas" mucho más pequeñas que los eritrocitos que ocasionalmente se agrupaban y participaban en colecciones defibrina, recomendando estudios adicionales sobre estos hallazgos.
Giulio Bizzozero (1846-1901), aportó sobre los hallazgos de Schultze, usando "circulación en vivo" para estudiar las células sanguíneas de anfibios microscópicamente. Él notó especialmente que las plaquetas se agrupaban en el sitio de lesión vascular, un proceso que precedía a la formación de un coágulo. Esta observación confirmó el papel de las plaquetas en la coagulación.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarCINÉTICA
Las plaquetas son producidas en el proceso de formación de las células sanguíneas llamado (trombopoyesis) en la médula ósea, por fragmentación en los bordes citoplasmáticos de los megacariocitos.
• El rango fisiológico de las plaquetas es de 150-400 x 109/litro.
• Alrededor de 1 x 1011 plaquetas de media son producidas cada día por un adulto sano.
• La expectativa de vida de las plaquetas circulantes es de 7 a 10 días.
• La producción de megacariocitos y plaquetas está regulada por la trombopoyetina, una hormona producida usualmente por el hígado y los riñones.
• Cada megacariocito produce entre 5.000 y 10.000 plaquetas.
• Las plaquetas son destruidas por fagocitosis en el bazo y por las células de Kupffer en el hígado.
• Una reserva de plaquetas es almacenada en el bazo y son liberadas cuando se necesitan por medio de contracción esplénica mediada por el sistema nervioso simpático.
Formación de trombos
La función plaquetaria consiste en el mantenimiento del corazón; Esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarACTIVACIÓN DE LAS PLAQUETAS
La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP.
Las células endoteliales producen una proteína llamada factor de von Willebrand (FvW),un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas.
Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo.
Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio.
La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria(plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina yfosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
CAMBIO DE FORMA
Las plaquetas activadas cambian su forma haciéndose más esféricas, y formando pseudopodos en su superficie. De esta forma toman una forma estrellada.
SECRECIÓN DE GRÁNULOS
Las plaquetas contienen gránulos alfa y gránulos densos. Las plaquetas activadas excretan el contenido de estos gránulos dentro de sus sistemas canaliculares y en la sangre circundante. Existen dos tipos de gránulos:
• Gránulos densos (contienen ADP o ATP, calcio, y serotonina)
• Granulos-α (contienen factor 4 plaquetario, factor de crecimiento transformante beta 1 (TGF beta 1), factor de crecimiento derivado de plaquetas, fibronectina, B-tromboglobulina, FvW, fibrinógeno, y factores de coagulación factor V y XIII).
SÍNTESIS DE TROMBOXANO A2
La activación plaquetaria inicia la vía del ácido araquidónico para producir Tromboxano A2; el tromboxano A2 está involucrado en la activación de otras plaquetas y su formación es inhibida por los inhibidores de la COX, como el ácido acetilsalicílico.
ADHESIÓN Y AGREGACIÓN
La agregación plaquetaria, usa el fibrinógeno y el FvW como agentes conectores. El receptor de agregación plaquetaria más abundante es la glicoproteina IIb/IIIa (gpIIb/IIIa); se trata de un receptor para el fibrinógeno dependiente del calcio, fibronectina, vitronectina,trombospondina, y factor de von Willebrand (FvW). Otros receptores incluyen el complejo GPIb-V-IX (FvW) y GPVI (colágeno).ó Las plaquetas activadas se adherirán, via glicoproteína (GP) Ia, al colágeno expuesto por el daño epitelial. La agregación y adhesión actúan juntos para formar el tapón plaquetario. Los filamentos de Miosina y actina en las plaquetas son estimuladas para contraerse durante la agregación, reforzando todavía más el tapón.
La agregación plaquetaria es estimulada por el ADP, tromboxano, y la activación del receptor-α2, pero inhibido por agentes antiinflamatorios como las prostaglandinas PGI2 y PGD2. La agregación plaquetaria se ve aumentada por la administración exógena de esteroídes anabólicos.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarREPARACIÓN DE HERIDAS
El coágulo sanguíneo es solo una solución temporal para detener la hemorragia; la reparación del vaso debe ocurrir después. La agregación plaquetaria ayuda en este proceso mediante la secreción de sustancias químicas que promueven la invasión de fibroblastosdel tejido conectivo adyacente hacia el interior de la herida para formar una costra. El coágulo obturador es lentamente disuelto por la enzima fibrinolítica, plasmina, y las plaquetas son eliminadas por fagocitosis.
Otras funciones
• Retracción del coágulo
• Pro-coagulación
• Inflamación
• Señalización citoquínica
• Fagocitosis
SEÑALIZACIÓN CITOQUÍNICA
Adicionalmente a su función de ser el efector celular de la hemostasia, las plaquetas son rápidamente depositadas en sitios de lesión o infección, y potencialmente modulan los procesos inflamatorios por medio de la interacción con leucocitos y por la secreción decitoquinas, quimiosinas, y otros mediadores de la inflamación .las plaquetas también secretan factor de crecimiento derivado de plaquetas (PDGF).
PAPEL EN ENFERMEDADES
Recuentos altos y bajos
El recuento de plaquetas de un individuo sano se encuentra entre 150,000 y 450,000 por μl (microlitro) de sangre (150-450 x 109/L).18 El 95 % de los individuos sanos tendrán recuentos de plaquetas dentro de este rango. Algunos tendrán recuentos de plaquetas estadísticamente anormales sin tener ninguna anormalidad demostrable. Sin embargo, si el recuento es muy alto o muy bajo la probabilidad de que una anormalidad este presente es más alta.
Tanto la trombocitopenia como la trombocitosis pueden manifestarse como problemas de coagulación. En general, los recuentos bajos de plaquetas incrementan el riesgo de sangrado; sin embargo existen excepciones. Por ejemplo la trombocitopenia inmune inducida por heparina.En la trombocitosis se puede producir trombosis, sin embargo esto sucede principalmente cuando el recuento elevado es debido a desordenes mieloproliferativos.
Los recuentos de plaquetas en general, no son corregidos con transfusión a menos que el paciente esté sangrando o el recuento haya caído por debajo 5 x 109/L. La transfusión esta contraindicada en la púrpura trombocitopénica idiopática (PTI), puesto que estimula lacoagulopatía. En los pacientes sometidos a cirugía, niveles inferiores a 50 x 109/L están asociados a sangrado quirúrgico anormal, y procedimientos anestésicos regionales como la anestesia epidural son evitados para niveles inferiores a 80-100 x 109/L.
El recuento normal de plaquetas no es garantía de función adecuada. En algunos estados, las plaquetas, siendo normales en número, son disfuncionales. Por ejemplo, el ácido acetilsalicílico interrumpe irreversiblemente la función plaquetaria mediante la inhibición de laciclooxigenasa-1 (COX1), y por consiguiente la hemostasia normal. Las plaquetas resultantes no tienen ADN y son incapaces de producir nueva ciclooxigenasa. La función plaquetaria normal no se restaurara hasta que el uso de ASA haya cesado y un número suficiente de las plaquetas afectadas hayan sido reemplazadas por nuevas, lo cual suele tardar unos 7 días. El Ibuprofeno, un AINE, no tiene un período tan largo de efecto, y la función plaquetaria vuelve a la normalidad dentro de las 24 horas,19 y tomando ibuprofeno antes que el ASA prevendrá los efectos irreversibles de esta.20 La Uremia, a consecuencia de la insuficiencia renal, conduce a la disfunción plaquetaria que puede ser aminorada con la administración de desmopresina.
MEDICAMENTOS
Agentes orales usados a menudo para alterar/suprimir la función plaquetaria:
• Ácido acetilsalicílico
• Clopidogrel
• Cilostazol
• Ticlopidina
Agentes intravenosos usados a menudo para alterar/suprimir la función plaquetaria:
• Abciximab
• Eptifibatida
• Tirofiban
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarENFERMEDADES
Desordenes que provocan un recuento bajo de plaquetas:
• Trombocitopenia
• Púrpura trombocitopénica idiopática
• Púrpura trombocitopénica trombótica
• Púrpura trombocitopenica inducida por medicamentos
• Enfermedad de Gaucher
• Anemia aplásica
Trastornos Aloimunes
• Trombocitopenia fetomaterna autoinmune
• Algunas reacciones trasfusionales
Desordenes que provocan disfunción o recuento reducido:
• Síndrome HELLP
• Síndrome urémico hemolítico
• Quimioterapia
• Dengue
• Deficiencia del almacenamiento en gránulos Alfa-Delta; es un desorden hemorrágico hereditario.21
Desordenes caracterizados por recuentos elevados:
• Trombocitosis, incluyendo trombocitosis esencial (recuento elevado, ya sea reactivo o como una expresión de trastorno mieloproliferativo); puede mostrar plaquetas disfuncionales.
Desordenes de la agregación y adherencia plaquetarios:
• Síndrome de Bernard-Soulier
• Tromboastenia de Glanzmann
• Síndrome de Scott's
• Enfermedad de von Willebrand
• Síndrome de Hermansky-Pudlak
• Síndrome de plaquetas grises
Desordenes del metabolismo de plaquetas
• Actividad disminuida de la oxigenación, inducida o congénita
• Defectos del almacenamiento, adquirido o congénito
Desordenes que comprometen la función plaquetaria:
• Hemofilia
Desordenes en los cuales las plaquetas juegan un papel clave:
• Aterosclerosis
• Enfermedad coronaria arterial, e infarto del miocardio
• Accidente cerebro vascular
• Enfermedad arterial oclusiva periférica
• Cancer
PLASMA RICO EN PLAQUETAS (PRP)
La preparación del plasma rico en plaquetas (PRP) de procedencia autóloga, requiere la extracción y recolección de sangre periférica del paciente, la separación de las plaquetas y el plasma de los otros elementos formes sanguíneos, y la posterior polimerización de la fibrina de dicho plasma para concentrar las plaquetas formando un gel rico en plaquetas con suficiente estabilidad como para ser implantado quirúrgicamente. Actualmente, algunos métodos comerciales para la preparación del PRP utilizan calcio y trombina bovina o bien, trombina preparada de forma autóloga para crear una matriz rica en plaquetas y fibrina (PRFM del inglés- platelet-rich fibrin matrix). La preparación de trombina autóloga requiere tiempo y pasos adicionales, así como un mayor volumen de sangre; por otro lado, el uso de trombina bovina ha sido asociado con el desarrollo de anticuerpos contra los factores de coagulación V y XI, y la misma trombina, aumentando de esta forma el riesgo de anormalidades en la coagulación. Adicionalmente, para asegurar una degranulación de las plaquetas y la formación de un coágulo estable, se utilizan grandes cantidades de trombina, esto puede causar una liberación inmediata de los factores de crecimiento.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarLa liberación de factores de crecimiento es desencadenada por la activación de las plaquetas, esta puede ser iniciada por una gran variedad de sustancias o estímulos como la trombina, el cloruro de calcio, el colágeno o el adenosina 5c-difosfato.Un coágulo sanguíneo de PRP contiene aproximadamente un 4% de glóbulos rojos, 95% de plaquetas y 1% de glóbulos blancos. Las propiedades del PRP están basadas en los múltiples factores de crecimiento y de diferenciación producidos y liberados a raíz de la activación de las plaquetas. Estos factores son críticos para la regulación y estimulación del proceso curativo de lesiones. Por otro lado, existe una segunda generación del concentrado de plaquetas que recibe el nombre de matriz rica en plaquetas y fibrina (PRFM del inglés- platelet-rich fibrin matrix), la cual es un mejoramiento del PRP preparado tradicionalmente.
Existe un método actual que evita el uso de trombina como activador. Este sistema utiliza únicamente calcio y centrifugación para activar la polimerización de la fibrina y formar así el PRFM. PRFM, en forma de gel o una membrana densa y flexible, puede ser aplicada al paciente y la liberación de los factores de crecimiento es desencadenada por los activadores autólogos presentes en el sitio de aplicación. Este método permite una liberación gradual de los factores de crecimiento en el sitio de aplicación, que pueden emitir señales a diferentes tipos celulares para que emitan una respuesta en momentos apropiados. Estudios in vitro indican que el PRFM presenta una liberación gradual y estable de los factores de crecimiento a lo largo de 7 días.
El plasma rico en plaquetas (PRP) puede obtenerse por medio de diferentes técnicas ya sean separadores celulares de propósitos generales o bien, separadores celulares para la concentración de plaquetas. Muchos productos comerciales se encuentran disponibles en este campo, la mayoría de ellos obtienen resultados similares, cuyas diferencias se deben fundamentalmente al precio, tiempo, espacio requerido y la tecnología necesaria para fabricarlo. Son pocos los productos comerciales disponibles para la obtención de una matriz rica en plaquetas y fibrina como producto final.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarCOAGULACIÓN
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdaderocambio de estado.
Este proceso es debido, en última instancia, a que unaproteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregadosmacromoleculares en forma de una red tridimensional.
El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimáticopor el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse.
Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas.
Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
FACTORES DE COAGULACIÓN
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc.
En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares.
A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenossintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarETAPAS DE LA CASCADA DE COAGULACIÓN
La cascada de coagulación se divide para su estudio, clásicamente en tres vias: La via intrínseca, la vía extrínseca y la vía común.
Las vías intrínseca y extrínseca son las vías de iniciación de la cascada, mientras que la vía común es hacia donde confluyen las otras dos desembocando en la conversión de fibrinógeno en fibrina.
Esta división es un tanto arbitraria y tiene más que ver con las deficiencias de las técnicas que en su momento se utilizaron para desentrañar los mecanismos implicados, que con lo que ocurre realmente en una lesión vascular; ya que en este último caso se establecen varias interrelacciones entre las vías de iniciación.
Mecanismo básico
Cada reacción de estas vías da como resultado el ensamblado de un complejo compuesto por una enzima (factor de coagulación activado), un sustrato (proenzima de un factor de coagulación) y un cofactor que actúa posibilitando la reacción.
Estos componentes se ensamblan en general sobre una superficiefosfolipídica y se mantienen unidos por medio de puentes formados por iones Ca2+. Por lo tanto la reacción en cascada tiende a producirse en un sitio donde este ensamblaje puede ocurrir; por ejemplo sobre la superficie de plaquetas activadas.
Tanto la vía intrínseca como la vía extrínseca desembocan en la conversión del factor X en Xa (la letra "a" como subíndice "a" significa "activado") punto en el que se inicia la vía común.
Vía intrínseca
Recibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso hemostático.
El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular.
En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación.
En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como lacelulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción.
A esta vía es posible subdividirla en tres pasos:
Formación del factor XIa
En esta etapa participan cuatro proteínas: Precalicreína, Quininógeno de alto peso molecular (HMWK) y los factores XII y XI. Esta etapa no requiere de iones calcio.
Estos cuatro factores se adsorben sobre la superficie cargada negativamente, formando el complejo cebador o de iniciación. De estos factores el XII funciona como verdadero iniciador, ya que si bien es una proenzima, posee una pequeña actividad catalítica que alcanza para activar a la precalicreína convirtiéndola en calicreína.
En segunda instancia la calicreína actúa catalíticamente sobre el factor XII para convertirlo en XIIa, una enzima muchísimo más activa. La actividad catalítica de la calicreína se ve potenciada por el HMWK.
Por último la proteasa XIIa actúa sobre el factor XI para liberar XIa.
Formación del factor IXa
El factor IX se encuentra en el plasma como una proenzima. En presencia de iones Ca2+ el factor XIa cataliza la ruptura de una unión peptídica en la molécula del factor IX para formar un glucopéptido de 10 KDa y liberar por otro lado al factor IXa.
El factor IX se encuentra ausente en personas con hemofilia tipo B.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarFormación del factor Xa
Sobre la membrana de las plaquetas se forma un complejo constituido por los factores IXa, X y VIII.
Los residuos gamma-carboxiglutamato de los factores IXa y X actúan como quelantes del ion Ca2+, permitiendo que estos componentes formen un complejo unido por medio de puentes de iones calcio y ayudando a que el complejo se ancle a los fosfolípidos de membrana.
Primero se unen los factores X y IXa a la membrana y luego se une el VIII.
El factor VIII es en realidad un homorodímero, formado por cuatro cadenas proteicas, cada una codificada por un gen diferente (VIII:C y VIII:R). El componente VIII:C es conocido como "componente antihemofílico" y actúa como cofactor del IXa en la activación del factor X, el componente VIII:R es el que permite la unión del factor VIII al complejo.
La ausencia del componente antihemofílico causa hemofilia A.
El complejo formado por los factores IXa-X-VIII-Fosfolípidos y Ca2+ actúa sobre el factor X para convertirlo en Xa. En este punto concluye la vía intrínseca.
Vía extrínseca
Recibió este nombre debido a que fue posible notar desde un primer momento que la iniciación de esta vía requería de factores ajenos a la sangre.
Cuando la sangre entra en contacto con tejidos lesionados o se mezcla con extractos de tejidos, se genera muy rápidamente factor Xa. En este caso la activación de la proenzima X es mediada por un complejo formado por factor VII, Ca2+ y factor tisular unido afosfolípidos provenientes de las membranas celulares rotas y de las plaquetas (antiguamente este complejo factor tisular-fosfolípidos era conocido como tromboplastina).
El factor tisular es una lipoproteína sintetizada en el endotelio de los vasos sanguíneos de todos los tejidos, aunque es especialmente abundante en pulmón, cerebro y placenta. El factor tisular se encuentra normalmente "secuestrado" en el interior de las células endoteliales y es secretado en respuesta a una lesión, o bajo el efecto de algunas citoquinas tales como el Factor de Necrosis Tumoral (TNF), InterLeucina 1 (IL-1); o por endotoxinas bacterianas.
La vía extrínseca es muy rápida, se cumple en apenas unos segundos y comprende dos pasos; mientras que la intrínseca insume varios minutos.
Formación del factor VIIa
En primera instancia el factor VII se une a la porción fosfolipídica del factor tisular gracias a sus residuos gamma-carboxiglutamato, utilizando iones Ca2+ como puentes. Este complejo provoca la activación del factor VIIa.
Formación del factor Xa
El complejo VIIa-III-Ca2+ actúa sobre el factor X convirtiéndolo en la proteasa activa Xa. En este punto termina la vía extrínseca y se inicia la vía común
Vía común
Llegando al punto en que se activa el factor X, ambas vías confluyen en la llamada vía común.
La vía común termina con la conversión de fibrinógeno en fibrina, y el posterior entrecruzamiento de la misma estabilizando el coágulo.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarLa vía común implica tres etapas:
Formación de trombina
La trombina (también llamada factor II a) es una proteasa generada por la ruptura de la cadena proteica de la proenzima protrombina (factor II), una glicoproteína constituida por 582 aminoácidos y con 12 puentes disulfuro intracatenarios.
La trombina se activa luego de que la proteasa Xa hidroliza dos uniones peptídicas de la protrombina. La Xa produce en primer término la escisión de un fragmento de 32 KDa de la región N-terminal de la cadena, cortándola sobre una unión arginina-treonina. En segundo término produce la ruptura de un enlace entre una arginina y una isoleucina; sin embargo estos dos últimos fragmentos permanecen unidos por un puente disulfuro.
La trombina es una serina-proteasa similar a la tripsina, pero mucho más selectiva. Ataca casi de manera exclusiva las uniones arginina con un aminoácido cargado positivamente en sus sustratos.
La conversión de protrombina a trombina debida al factor Xa se acelera notablemente por la formación de un complejo con el factor Va y Ca2+ sobre la superficie de las membranas plaquetarias (fosfolípidos de membrana).
El factor Xa y la protrombina se adsorben sobre la membrana utilizando iones Ca2+como puentes. El factor Va se une a la protrombina acelerando la reacción.
El factor Va se produce por la acción de la trombina sobre el factor V en un claro ejemplo de una reacción que va acelerándose a medida que progresa (reacción autoacelerada).
Formación de fibrina
El fibrinógeno (factor I) es una glicoproteína compuesta por seis cadenas polipeptídicas: dos A-alfa, dos B-beta y dos gamma; unidas entre sí por puentes disulfuro.
Se trata de una molécula alargada y simétrica formada por tres dominios globulares conectados por segmentos fibrilares.
Cada mitad de la molécula se encuentra formada por tres cadenas (A-alfa, B-beta y gamma) que se enrollan en una triple hélice muy compacta en los sectores fibrilares. Los extremos amino de las seis cadenas se reúnen en el dominio globular central.
En un hecho que parecería muy curioso, los extremos N-terminales de las cadenas A-alfa y B-beta emergen como cabos libres del dominio globular central.
Estas cadenas son muy ricas en aspartato y glutamato, además las cadenas B-beta poseeen en esta región residuos tirosina-O-sulfato formados postraduccionalmente. Estos residuos con una alta tendencia a adquirir carga negativa contribuyen a formar una región central con una muy alta densidad de carga.
Esta región electronegativa central es la responsable de la repulsión entre moléculas de fibrina que las mantiene en solución.
La trombina ataca los enlaces arginina-glicina presentes en estos "cabos libres", separando cuatro péptidos; dos segmentos A de 18 aminoácidos cada uno (provenientes de las cadenas A-alfa), y dos segmentos B de 20 aminoácidos (provenientes de las cadenas B-beta). A estos péptidos se los suele denominar "fibrinopéptidos".
El resto que queda de la molécula es un monómero de fibrina de composición alfa2beta2gamma2.
Al eliminarse los fibinopéptidos desaparecen las fuerzas de repulsión intermoleculares con lo que los monómeros de fibrina tienden a agruparse espontáneamente formando asociaciones altamente ordenadas.
Los monómeros se disponen uno a continuación del otro, cabeza con cabeza en forma de largas hebras. Estas hebras a su vez forman manojos, emparejándose con otras hebras de tal manera que la región central de los monómeros de fibrina de una se encuentra rodeada por las cabezas de los monómeros de fibrina de las otras.
Este emparejamiento se hace posible gracias a interaciones de tipo electrostático y puente hidrógeno entre las regiones centrales de los monómeros de una y las cabezas globulares de otras.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarEntrecruzamiento de la fibrina
Los haces paralelos de fibrina polimerizada forman una asociación laxa, que se encuentra en equilibrio con la forma monomérica de la molécula; por lo que sería imposible que cumplieran su papel de formar un coágulo estable sin reforzar esta estructura por medio de enlaces covalentes entre hebras vecinas.
La formación de estos "puentes" covalentes intercatenarios es catalizada por la enzima transglutaminasa (conocida también como factor XIIIa).
La transglutaminidasa cataliza la formación de enlaces amida entre restos glutamina y lisina de hebras próximas entre sí. En la reacción se libera amoníaco en forma de ion amonio (NH4+).
Esta enzima se forma a partir del factor XIII por acción de la trombina.
Regulación y modulación de la cascada
Debido a que la cascada de coagulación consiste en una serie de reacciones que van amplificándose y acelerándose en cada paso, es lógico pensar que debe existir algún mecanismo de regulación; un "freno" a la reacción en cadena; ya que de progresar sin control en pocos minutos podría provocar un taponamiento masivo de los vasos sanguíneos (trombosis diseminada).
Varios mecanismos intervienen en la regulación de la cascada de reacciones:
• El flujo sanguíneo normal, arrastra a los factores activados, diluyendo su acción e impidiéndoles acelerarse. Esta es una de las razones por las cuales cuando existe estasis del flujo sanguíneo se favorece la formación de trombos.
• El hígado actúa como un filtro quitando de la sangre en circulación los factores activados e inactivándolos.
• Existen además algunas proteasas que degradan específicamente a ciertos factores activados, y otras que ejercen acciones inhibitorias sobre factores activos.
Proteína C
La proteína C es una proenzima que se encuentra normalmente en el plasma, y cuya síntesis en el hígado es dependiente de la vitamina K.
Esta proteína es convertida en una proteasa activa por la acción de la trombina.
La proteína Ca actúa específicamente degradando a los factores Va y VIIIa, con lo que limita la proyección de la cascada.
Es interesante notar el triple papel que desempeña la trombina: cataliza la formación de fibrina, activa a la enzima responsable de su entrecruzamiento, y una vez que el proceso de coagulación y estabilización del coágulo está en marcha; ejerce acciones tendientes a limitarlo.
Antitrombina III
La antitrombina III es una glicoproteína de 60 Kda sintetizada en el hígado sin depender de la vitamina K, es considerada la principal inhibidora de la coagulación.
Esta proteína actúa inhibiendo irreversiblemente a varios factores procoagulantes activos, el principal de los cuales es la trombina; aunque también actúa sobre la calicreína y los factores IXa, Xa, XIa y XIIa.
La acción de la antitrombina es notablemente aumentada por el heteropolisacárido heparina. La heparina se encuentra en el endotelio de los vasos sanguíneos y en los gránulos de las células cebadas, tiene una poderosa acción anticoagulante ya que facilita la unión de la antitrombina III con los factores procoagulantes activos.
Existen otras anti-proteasas sanguíneas que también ejercen acción anticoagulante aunque menos potente tales como la alfa2macroglobulina y la alfa1 antitripsina.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarAnticoagulantes
Un anticoagulante es, como su nombre lo indica, una sustancia química que retrasa o impide la coagulación de la sangre, ya sea en el interior de un organismo (In Vivo) o en el exterior (In Vitro)
Existen diferentes tipos de anticoagulantes que actúan dificultando o impidiendo alguno de los pasos de la cascada de coagulación.
Existen dos tipos principales de anticoagulantes, los anticoagulantes para uso "In Vitro" y los que tienen empleo "In Vivo", entre estos últimos se encuentran los medicamentos con acción anticoagulante.
En general los anticoagulantes para uso In Vitro actúan como quelantes del ion Ca2+, de manera tal que este no puede participar en la formación de los complejos que activan al factor X, y por lo tanto se interrumpe la cascada de coagulación casi en su inicio.
Los anticoagulantes para uso In Vivo actúan de maneras un poco más complicadas. La adición de algunos agentes quelantes tales como el EDTA entrañan un grave riesgo para la salud del individuo sometido a tratamiento, ya que estos agentes son capaces de acomplejar gran cantidad de iones con alta afinidad, algunos de los cuales desempeñan importantes funciones en el organismo tales como el Cu2+, Fe3+, Zn2+, etc; mientras que otros agentes acomplejantes del calcio tales como el citrato, no tienen gran utilidad ya que son rápidamente metabolizados perdiendo su capacidad anticoagulante.
Entre los anticoagulantes para uso in vivo encontramos sustancias tales como la heparina o los anticoagulantes dicumarínicos.
Para uso In Vitro
EDTA (C10H16N2O8) o sal disódica, dipotásica o tripotásica del ácido etilendiaminotetraacético. Esta sustancia actúa mediante un efecto quelante sobre el ion calcio (Ca2+, lo que impide la formación de los complejos procoagulantes en los que este ion participa. Este anticoagulante se utiliza fundamentalmente para la realización de recuentos celulares, sobre todo en autoanalizador. Tiene la ventaja de permitir la realización del hematocrito y de frotis sanguíneo hasta dos horas después de la extracción de la muestra. También impide la aglutinación de las plaquetas.
• Heparina Sódica. Heparina de Litio, es un anticoagulante fisiológico que actúa impidiendo que la protrombina se transforme en trombina. Estructuralmente es un mucopolisacárido ácido que posee grupos sulfato. Esta última característica no la hace adecuada para muestras que van a ser examinadas al microscopio luego de tinción, ya que altera notablemente las coloraciones obtenidas.
• Citrato Trisódico (C6H5O7Na3) actúa impidiendo que el calcio se ionice, evitando así la coagulación. Se utiliza principalmente para realizar pruebas de hemostasia; así como también para medir la velocidad de eritrosedimentación.
• ACD, es un anticoagulante formado por una mezcla de compuestos (Ácido citrico Citrato y Dextrosa en una proporción de 0.9, 2 y 2 g respectivamente en 120 ml de agua destilada) se emplea fundamentalmente en bancos de sangre para conservar las unidades de sangre y para realizar estudios metabólicos eritrocitarios ya que permite una buena conservación de los hematíes.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarAnticoagulantes para uso In Vivo (medicamentos anticoagulantes)
Este grupo de anticoagulantes se definen como "medicamentos que impiden la coagulación o la agregación plaquetaria"
Este tipo de medicamentos tienen utilidad en aquellas patologas causadas por un trombo sanguíneo, ya sea para facilitar su disolución (trombolisis) o bien para prevenir que los trombos se repitan.
En este artículo vamos a centrarnos en aquellos que impiden la cascada de coagulación:
• La heparina alarga el tiempo de coagulación, se administra generalmente mediante inyección subcutánea o endovenosa.
Ya que es un compuesto fisiológico presente en gran cantidad en los mamíferos, comúnmente se utiliza heparina obtenida de pulmón de vaca o de mucosa intestinal de cerdo convenientemente purificada. La potencia difiere según el origen, pero hoy en día vienen estandarizadas en UI, por lo que se pueden comparar solo con este índice.
Comercialmente se obtiene en forma de dos sales (cálcica y sódica) que no guardan demasiada diferencia en su actividad. Las cálcicas se usan preferentemente por vía subcutánea, ya que resultan menos dolorosas, pero por vía endovenosa pueden utilizarse ambas. La heparina NUNCA se administra vía intramuscular.
La Heparina se utiliza cuando se precisa de acción anticoagulante rápida y por poco tiempo. En la prevención de trombosis venosas de cirugía se utiliza a bajas dosis, 5.000UI, dos horas antes de la intervención y después cada 12 horas hasta el alta del paciente.
Las heparinas de bajo peso molecular son fragmentos de peso molecular entre 3.500 y 6.000, con ello tiene una vida más larga y aumenta su biodisponibilidad. Tiene una menor inhibición de la agregación plaquetaria. No sustituyen a las heparinas tradicionales sino que en terapias de baja dosis son más cómodas porque se aplican una sola vez al día.
En terapias de altas dosis se utilizan las heparinas tradicionales.
• Hirudina
• Anticoagulantes dicumarínicos. Reciben este nombre genérico un grupo de compuestos derivados del Dicumarol (un compuesto extraído del trébol dulce) entre los que se encuentran el Acenocumarol (el de uso más frecuente en España, bajo el popular nombre de Sintrom) y la Warfarina. Estos medicamentos presentan la ventaja de poder ser administrados por vía oral y de poseer un efecto prolongado en el tiempo, con gran variabilidad interindividual, por ello necesitan controles periódicos para su ajuste terapéutico.
Todos ellos son inhibidores de la vitamina K (aVK). Debido a que la vitamina K interviene como cofactor enzimático en la síntesis de los factores II,VII,IX y X (concretamente en la gamma-carboxilación de estos); el resultado es que provoca la aparición en sangre, de unas formas inactivas de los mismos denominadas PIVKAs (“Proteins Induced by Vitamin K Antagonists”).
Dada la diferente vida media que presentan los factores de coagulación (el tiempo que permanecen en sangre antes de ser degradados), por ejemplo el VII comienza a descender en 6 horas pero el II tarda cerca de 70, no se consigue una anticoagulación efectiva hasta el 3º-4º día de tratamiento y el efecto no se estabiliza hasta después de una semana.
Curioso es que la activación de dos inhibidores fisiológicos de la coagulación como son las Proteínas C y S de importancia fundamental (inhiben a los Factores V y VIII activados), también depende de la Vit. K, por lo que los cumarínicos originan una “paradoja bioquímica” anticoagulante-procoagulante.
No obstante, su efecto anticoagulante supera ampliamente al procoagulante, por lo que solo puede tener consecuencias clínicamente significativas en raros casos (Déficits congénitos de Proteína C o S) y de forma transitoria al inicio del tratamiento.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarTROMBOS Y COÁGULOS
Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida.
• Un coágulo sanguíneo que se forma dentro de una de las venas o las arterias se denomina trombo y también se puede formar en el corazón.
• Un trombo que se desprende y viaja desde un lugar en el cuerpo a otro se llama émbolo.
Un trombo o émbolo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo.
• Una obstrucción en una arteria puede impedir que el oxígeno llegue a los tejidos en esa área, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede provocar daños en los tejidos o la muerte.
• Una obstrucción en una vena generalmente provocará acumulación de líquido e hinchazón.
Causas
Las situaciones en las que es más probable que se forme un coágulo de sangre abarcan:
• Estar en reposo en cama por largo tiempo.
• Cruzar las piernas durante largos periodos al estar sentado o sentarse durante mucho tiempo, como en un avión o en un vehículo.
• Durante y después del embarazo.
• No tener suficiente agua en el cuerpo (deshidratación).
• Tomar píldoras anticonceptivas u hormonas estrógeno (especialmente en las mujeres que fuman).
• Utilizar un catéter intravenoso por largo tiempo.
Los coágulos de sangre también son más probables en personas con cáncer, cirugía o lesión recientes, obesidad y enfermedades del hígado o del riñón.
Una acumulación de colesterol que estreche una arteria puede cambiar o reducir el flujo de sangre, facilitando la formación de un coágulo sanguíneo o de un trombo.
Las afecciones que se transmiten de padres a hijos (hereditarias) pueden hacerlo más propenso a la formación de coágulos sanguíneos anormales. Los trastornos hereditarios que afectan la coagulación son:
• Trombofilia por factor V de Leiden
• Mutación de la protrombina G20210A
• Otras afecciones raras como deficiencias de antitrombina III, proteína C y S
Un coágulo de sangre puede bloquear una arteria o una vena en el corazón, afectando:
• El corazón (angina de pecho o un ataque cardíaco)
• Los intestinos (isquemia mesentérica) o trombosis venosa mesentérica
• Los riñones (trombosis de la vena renal)
• Las arterias de las piernas o brazos
• Las piernas (trombosis venosa profunda)
• Los pulmones (embolia pulmonar)
• El cuello o el cerebro (accidente cerebrovascular)
Posibles complicaciones
Los trombos y émbolos pueden fijarse firmemente a un vaso sanguíneo y pueden bloquear parcial o completamente el flujo de sangre a dicho vaso.
Un bloqueo en el vaso sanguíneo impide que el flujo normal de sangre y oxígeno llegue a los tejidos en ese sitio, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede ocasionar daño tisular o necrosis en esa área.
Nelly Suleyka Solano Rodriguez 86814
ResponderEliminarFIBRINÓLISIS
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos.
Bioquímica
La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasasen los macrófagos del hígado y el riñón.
La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca:
• En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o lasinfecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógenoo tPA.
• En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno.
Regulación
La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Después de que el coágulo se ha establecido, comienza la reparación de los tejidos afectados con el proceso de cicatrización. Para hacer posible esto el coágulo es colonizado por células que formarán nuevos tejidos y en el proceso va siendo degradado.
La degradación de la fibrina (fibrinólisis), componente mayoritaria del coágulo, es catalizada por la enzima plasmina, una serina proteasa que ataca las uniones peptídicas en la región triple hélice de los monómeros de fibrina.
La plasmina se genera a partir del plasminógeno, un precursor inactivo; activándose tanto por la acción de factores intrínsecos (propios de la cascada de coagulación) como extrínsecos, el más importante de los cuales es producido por el endotelio vascular. Se le denomina "activador tisular del plasminógeno" (t-PA).
El gen de este factor ha sido clonado y actualmente se puede obtener la proteína producida por tecnología de ADN recombinante.
Este factor suele utilizarse en clínica para favorecer la disolución de trombos.
SISTEMA FIBRINOLITICO
El sistema fibrinolítico va a jugar además un papel importante en diversas situaciones en las que se produce proteolisis tisular, tales como inflamación, invasión tumoral o neovascularización. La importancia fisiopatológica del sistema fibrinolítico deriva de que las alteraciones que condicionan un defecto de actividad fibrinolítica pueden predisponer a la trombosis, mientras que un exceso de activación favorecería la aparición de hemorragia. Además va a tener una gran importancia en el área terapéutica por la aplicación creciente de fármacos fibrinolíticos en el tratamiento de la trombosis, especialmente en el infarto agudo de miocardio (IAM).
Principales componentes del sistema fibrinolítico: La fibrinolisis es un encadenamiento (proceso) enzimático compuesto por una serie de activadores e inhibidores los cuales regulan la conversión del proenzima circulante, plasminógeno, en el enzima activo plasmina. La producción de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina y esto es muy importante para el mantenimiento de la permeabilidad vascular.
dilenny quezada 83876
ResponderEliminaractivacion plaquetaria
Los procesos de activación plaquetaria comprenden complejas rutas de señalización que involucran sistemas de proteínas G acoplados a enzimas efectoras como la fosfolipasa A2, fosfolipasa C, adenilato y guanilato ciclasas. Las propiedades funcionales y estructurales de las plaquetas están estrechamente relacionadas, lo que les confiere un alto potencial trombogénico, contribuyendo sustancialmente en el desarrollo de un infarto cerebral. Se realizó una revisión de las bases de la agregación, activación, degranulación plaquetaria y su participación en la enfermedad cerebrovascular. Se realizó una actualización del uso de los agentes antiplaquetarios y de la literatura actual referente al uso de bloqueadores de las glicoproteínas adhesivas de membrana.
Palabras clave: Activación plaquetaria, Infarto cerebral, Glicoproteína IIb/IIIa, Antiagregantes plaquetarios.
dilenny quezada 83876
ResponderEliminarcuagulacion de la sangre 1
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse.
Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas.
Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo
dilenny quezada 83876
ResponderEliminartrombo y cuagulo
Trombo: es una masa intravascular o intracardíaca formada por elementos de la sangre e insolubles en ella, se caracterizan por estar fijos a la pared del vaso por una zona donde son más rígidos y consistentes. Pueden ser arteriales o venosos.
Coagulos: intravasculares son postmorten son gelatinosos, con una porción declive rojo oscura porque los eritrocitos sedimentan por acción de la graverdad y un sobrenadante amarillo en grasa de pollo, no estan fijos a la pared del vaso.
En cambio los trombos son mas consistentes, estan anclados en un punto a la pared del vaso o cardiaca, los arteriales consistentes de color blanco grisaceo y friables, formados por una red de plaquetas fibrina y leucocitos en degeneración (se generan en zonas donde hay turbulencia, como en la bifircación de los vasos) lo venosos son rojos y se desarrollan en zonas de estasis sanguineo por lo que retienen mayor número de hematíes, por eso son rojos...
dilenny quezada 83876
ResponderEliminarfibrinolisis
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos
La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón.
La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca:
En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA.
En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno.
dilenny quezada 83876
ResponderEliminarsistema fibrinolitico
REGULACIÓN DEL SISTEMA FIBRINOLÍTICO
La hemostasia fisiológica asegura la permeabilidad vascular y la circulación sanguínea gracias a un complejo balance entre los mecanismos de coagulación, encargados de la formación de fibrina, y los de fibrinolisis responsables de su eliminación del torrente circulatorio. La fibrinolisis es un proceso específico de disolución de fibrina por proteasas sanguíneas, siendo la plasmina el enzima responsable de dicha degradación. La activación del sistema fibrinolítico es esencial para eliminar depósitos intravasculares de fibrina resultantes de la activación fisiológica o patológica del sistema de coagulación.
El sistema fibrinolítico va a jugar además un papel importante en diversas situaciones en las que se produce proteolisis tisular, tales como inflamación, invasión tumoral o neovascularización (3, 4). La importancia fisiopatológica del sistema fibrinolítico deriva de que las alteraciones que condicionan un defecto de actividad fibrinolítica pueden predisponer a la trombosis, mientras que un exceso de activación favorecería la aparición de hemorragia. Además va a tener una gran importancia en el área terapéutica por la aplicación creciente de fármacos fibrinolíticos en el tratamiento de la trombosis, especialmente en el infarto agudo de miocardio (IAM).
Principales componentes del sistema fibrinolítico: La fibrinolisis es un encadenamiento (proceso) enzimático compuesto por una serie de activadores e inhibidores los cuales regulan la conversión del proenzima circulante, plasminógeno, en el enzima activo plasmina. La producción de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina (5) y esto es muy importante para el mantenimiento de la permeabilidad vascular (Fig. 1). Los principales componentes del sistema fibrinolítico se resumen en la Tabla I.
mirian sujeidy marrero 85500
ResponderEliminaractivacion plaquetaria
El factor de activación plaquetaria es un fosfolípido de síntesis endógena relacionado directamente con diversos procesos como isquemia, necrosis, trombosis y otros. El principal blanco de sus efectos deletéreos se encuentra en las células endoteliales, leucocitos y plaquetas, donde provoca graves perturbaciones mecánicas, reológicas y bioquímicas en la unidad circulatoria. Se realizó una revisión bibliográfica del factor de activación plaquetaria, su síntesis endógena, sus funciones biológicas, con especial énfasis en su relación con el daño oxidativo, así como los principales antagonistas conocidos. Se enfatizó el efecto de los ginkgólidos contenidos en el EGB 761 (extracto de Ginkgo biloba) y su aplicación en la clínica de diversos procesos patológicos relacionados con la circulación cerebral y periférica.
mirian sujeidy marrero 85500
ResponderEliminarcuagulacion de la sangre 1
Son un grupo de afecciones en las cuales hay un problema con el proceso de coagulación sanguínea del cuerpo. Estos trastornos pueden llevar a que se presente sangrado intenso y prolongado después de una lesión. El sangrado también puede iniciarse de manera espontánea
Causas
La coagulación normal de la sangre involucra hasta 20 proteínas plasmáticas diferentes, conocidas como factores de la coagulación o factores de la coagulación sanguínea. Estos factores interactúan con otros químicos para formar una sustancia llamada fibrina que detiene el sangrado.
Se pueden presentar problemas cuando faltan ciertos factores de la coagulación o éstos están muy bajos. Los problemas de sangrado pueden ir desde leves hasta severos.
Algunos trastornos hemorrágicos están presentes desde el nacimiento y se transmiten de padres a hijos (hereditarios). Otros se desarrollan por:
•Enfermedades como deficiencia de vitamina K y enfermedad hepática severa
•Tratamientos como el uso de medicamentos para detener los coágulos de sangre (anticoagulantes) o el uso prolongado de antibióticos
mirian sujeidy marrero 85500
ResponderEliminarcuagulacion de la sangre 2
Es un problema de coagulación de la sangre que ocurre cuando hay una falta de una sustancia (protrombina) que se necesita para que la sangre coagule
Cuando uno sangra, el cuerpo inicia una serie de reacciones que ayudan a que la sangre se coagule, lo cual se denomina cascada de la coagulación. El proceso involucra proteínas especiales, llamadas factores de coagulación. Cuando falta uno o más de estos factores de coagulación, generalmente hay una probabilidad más alta de sangrado.
Este trastorno ocurre cuando el cuerpo no tiene suficiente factor II, una importante proteína en la coagulación de la sangre. La deficiencia del factor II que se transmite de padres a hijos (hereditaria) es muy poco frecuente. Ambos padres deben ser portadores para transmitírselo a sus hijos. El hecho de tener un antecedente familiar de un trastorno hemorrágico es un factor de riesgo potencial.
Con mucha frecuencia, la deficiencia del factor II es causada por:
Falta (deficiencia) de vitamina K por el uso prolongado de antibióticos, obstrucción de las vías biliares y absorción inadecuada de los nutrientes por parte del tracto intestinal. (malabsorción intestinal).
Enfermedad hepática grave.
Uso de medicamentos para prevenir la coagulación (anticoagulantes como warfarina o Coumadin).
examenes
Análisis del factor II
Tiempo parcial de tromboplastina
Tiempo de protrombina (TP)
mirian sujeidy marrero 85500
ResponderEliminarcuagulacion de la sangre 3
La Hemostasis es el proceso mediante el cual un organismo evita las pérdidas excesivas de sangre cuando hay daño en algún vaso sanguíneo. Una herida en el cuerpo causa vasoconstricción acompañada de la adhesión de plaquetas y la formación de fibrinas. La activación de las plaquetas conlleva la formación de un tapón haemostatico, que para el sangrado y después es reforzado por la fibirina. La importancia y velocidad de cada proceso depende del tipo de vena, arteria o capilar que haya sido lastimado. Los tapones haemostáicos en situaciones patológicas son conocidos como trombos y siempre ocurren in vivo, mientras que los coágulos son formaciones en sangre estática o in vitro.1
Cuadro
La Trombosis es la formación patológica de tapones hemostaticos en el sistema vascular sin la presencia de sangrado. Existen tres principales factores de disposición a la trombosis, que incluyen el daño a la pared vascular de un vaso sanguíneo o su erosión, un flujo sanguíneo alterado (ya sea por factores genéticos como mal funcionamientos o ambientales como el estilo de vida) o una coagulabilidad anormal de la sangre como sucede en las últimas fases del embarazo o mientras se lleva un tratamiento con anticonceptivos orales.2
La coagulación de la sangre es el proceso encargado de convertir la sangre en una sustancia gelatinosa. El evento principal es la conversión de fibrinógenos solubles a tiras insolubles de fibrinas mediante la acción de la trombina, y también es el último paso de esta cascada enzimática. Los componentes o factores de coagulación están siempre presentes en la sangre como precursores inactivos de enzimas proteolíticas y cofactores. Se activan mediante proteólisis, y su forma activa se denomina con el sufijo a. En esta cascada, la activación de una pequeña cantidad de factores cataliza la activación de grandes cantidades de otros y así sucesivamente, consecuentemente esta cascada provee un mecanismo de amplificación. La cascada está controlada con inhibidores para evitar la solidificación de toda la sangre del cuerpo. Uno de los inhibidores más importantes es la antitrombina III que neutraliza las proteasas serínicas de la cascada (XIa, Xa, IXa y IIa). El endotelio vascular también ayuda a limitar la zona de coagulación.1
Los factores de coagulación y sus funciones principales se muestran a continuació
mirian sujeidy marrero 85500
ResponderEliminartrombo y coagulo
El trombo es un coágulo sanguíneo que se forma en un vaso y permanece allí. La embolia es un coágulo que se desplaza desde el sitio donde se formó a otro lugar en el cuerpo. El trombo o embolia puede producirse en un vaso sanguíneo y obstruir el flujo sanguíneo en ese lugar, impidiendo el suministro de oxígeno y flujo sanguíneo a los tejidos circundantes. Esto puede ocasionar un daño, destrucción (infarto) e incluso la muerte o necrosis de los tejidos que se encuentran en esa área.
Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida.
Un coágulo sanguíneo que se forma dentro de una de las venas o las arterias se denomina trombo y también se puede formar en el corazón.
Un trombo que se desprende y viaja desde un lugar en el cuerpo a otro se llama émbolo.
Un trombo o émbolo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo.
Una obstrucción en una arteria puede impedir que el oxígeno llegue a los tejidos en esa área, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede provocar daños en los tejidos o la muerte.
Una obstrucción en una vena generalmente provocará acumulación de líquido e hinchazón.
Las situaciones en las que es más probable que se forme un coágulo de sangre abarcan:
Estar en reposo en cama por largo tiempo.
Cruzar las piernas durante largos periodos al estar sentado o sentarse durante mucho tiempo, como en un avión o en un vehículo.
Durante y después del embarazo.
No tener suficiente agua en el cuerpo (deshidratación).
Tomar píldoras anticonceptivas u hormonas estrógeno (especialmente en las mujeres que fuman).
Utilizar un catéter intravenoso por largo tiempo.
Los coágulos de sangre también son más probables en personas con cáncer, cirugía o lesión recientes, obesidad y enfermedades del hígado o del riñón.
mirian sujeidy marrero 85500
ResponderEliminarfibrinolisis
. Disolución de la fibrina y, por extensión, disolución de un coágulo sanguíneo (trombólisis). Es un fenómeno que sobreviene normalmente algunos días o algunas semanas después de la formación del coágulo. Cuando se produce demasiado rápidamente, puede provocar hemorragias espectaculares (fibrinólisis hemorrágica) intra o postoperatorias (particularmente en Cirugía Torácica), o después de un parto, de un aborto o de un choque traumático; o bien puede provocar hemorragias menos importantes (equimosis, hematuria) en el curso de ciertos cánceres (próstata, páncreas, estómago), de las cirrosis y de las leucosis. Obedece a la liberación de activadores de la profibrinolisina, o bien a la producción de un fermento proteolítico por los tejidos enfermos. Puede ser primitiva aunque por lo general es secundaria, reactiva, en el curso de un síndrome de coagulación intravascular diseminada
La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
LADY BAEZ 75261
ResponderEliminarActivacion plaquetaria
La sangre circula por el interior de los vasos, los glóbulos rojos (la más abundante masa celular sanguínea) lo hace por el centro del vaso, mientras que las plaquetas circulan más próximas a la superficie endotelial para vigilar la integridad del endotelio. El endotelio normal proporciona una superficie no adhesiva para las plaquetas. Cuando se rompe el endotelio, el colágeno expuesto favorece la adhesión de las plaquetas por medio de la interacción con una cantidad de receptores, como vemos en el video, pero es esencial para concretar la adhesión el Factor de Von Willebrand. A partir de esto y con el aumento del Ca++ intracelular se produce la activación de las plaquetas, lo que genera una serie de eventos que favorece la agregación plaquetaria, como son:
-Cambio de forma y de ser una esfera regular pasa a tener prolongaciones.
-Liberación del contenido de los gránulos (degranulación):
ADP que torna a las plaquetas pegajosas,TX A2 derivado del Ácido Araquidónico (vasoconstrictor y agreganteplaquetario)
actor II (trombina).
Adhesión, Activación y Agregación son las fases que participan en la formación del tapón plaquetario .
Proceso de coagulacion
Los factores de coagulación son proteínas de la sangre que controlan el sangrado.
Cuando un vaso sanguíneo se lesiona, sus paredes se contraen para limitar el flujo de sangre al área dañada. Entonces, pequeñas células llamadas plaquetas se adhieren al sitio de la lesión y se distribuyen a lo largo de la superficie del vaso sanguíneo. Al mismo tiempo, pequeños sacos al interior de las plaquetas liberan señales químicas para atraer a otras células al área y hacer que se aglutinen a fin de formar lo que se conoce como tapón plaquetario.
En la superficie de estas plaquetas activadas muchos factores de coagulación diferentes trabajan juntos en una serie de reacciones químicas complejas (conocidas como cascada de la coagulación) para formar un coágulo de fibrina. El coágulo funciona como una red para detener el sangrado.
Los factores de la coagulación circulan en la sangre sin estar activados. Cuando un vaso sanguíneo sufre una lesión se inicia la cascada de la coagulación y cada factor de la coagulación se activa en un orden específico para dar lugar a la formación del coágulo sanguíneo. Los factores de la coagulación se identifican con números romanos (e. g. factor I o FI).
Fibrinolisis
La fibrinólisis primaria se refiere a la descomposición normal de los coágulos, mientras que la fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa.
Consecuencias
Cuando se produce demasiado rápidamente, puede provocar hemorragias espectaculares (fibrinólisis hemorrágica) intra o postoperatorias (particularmente en Cirugía Torácica), o después de un parto, de un aborto o de un choque traumático; o bien puede provocar hemorragias menos importantes (hematuria) en el curso de ciertos cánceres (próstata, páncreas, estómago), de las cirrosis y de las leucosis. Obedece a la liberación de activadores de la profibrinolisina, o bien a la producción de un fermento proteolítico por los tejidos enfermos.
Nelsie Ortiz 84731
ResponderEliminarPlaquetas, coagulación y fribinolisis
1. Activación Plaquetaria: Los procesos de activación plaquetaria comprenden complejas rutas de señalización que involucran sistemas de proteínas G acoplados a enzimas efectoras como la fosfolipasa A2, fosfolipasa C, adenilato y guanilato ciclasas. Las propiedades funcionales y estructurales de las plaquetas están estrechamente relacionadas, lo que les confiere un alto potencial trombogénico, contribuyendo sustancialmente en el desarrollo de un infarto cerebral. Se realizó una revisión de las bases de la agregación, activación, degranulación plaquetaria y su participación en la enfermedad cerebrovascular. Se realizó una actualización del uso de los agentes antiplaquetarios y de la literatura actual referente al uso de bloqueadores de las glicoproteínas adhesivas de membrana.
2. Coagulación de la sangre 1, 2, 3 y 4: La coagulación de la sangre requiere plaquetas también denominadas trombocitos y ciertas proteínas llamadas factores de coagulación. Las plaquetas son células con forma de óvalo que se producen en la médula ósea. La mayoría de los factores de coagulación se generan en el hígado. Cuando los vasos sanguíneos se lesionan, las plaquetas son las primeras en llegar al área de la lesión para sellar la pérdida y reducir o detener temporariamente el sangrado. Pero para que el coágulo sea firme y estable, es necesario que intervengan los factores de coagulación.Los doce factores de coagulación del organismo se enumeran con números romanos la tromboplastina es el factor III. Estos factores actúan juntos en una secuencia especializada, casi como las piezas de un rompecabezas. El coágulo se forma al colocarse la última pieza. Pero si falta una pieza, o ésta es defectuosa, el coágulo no puede formarse.
3. Trombo y Cuagulo: Los coágulos sanguíneos o de fibrina son masas que se forman cuando la sangre se coagula y un trombo es un coágulo de sangre que se forma en un vaso o dentro del corazón y permanece allí. Un émbolo es un trombo que viaja desde el vaso o la cámara del corazón donde se formó a otro lugar del cuerpo y el trastorno causado se llama embolia o embolismo.
4. Fibrinolisis: Es un proceso corporal normal que impide que los coágulos sanguíneo que ocurren en forma natural crezcan y causen problemas. La fibrinólisis primaria se refiere a la descomposición normal de los coágulos. La fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa. Esto puede provocar sangrado intenso.
Av86070 Alejandra Valle:
ResponderEliminarYa que han abarcado tan bien los temas decidí buscar información relacionada a la agregación plaquetaria y estudios científicos relacionados a ella.
Efecto del ozono sobre la activación plaquetaria en pacientes diabéticos tratados con ozonoterapia:
Se verificó in vivo, en los pacientes con diagnóstico de pie diabético isquémico o neuroinfeccioso a los cuales se le indicó tratamiento con 200 mL de ozono, si existía inhibición o no de la agregación plaquetaria al final de este tratamiento (10 a 20 d). La agregación plaquetaria encontrada al inicio, antes del comienzo del tratamiento con ozono, tuvo una media de 28,3 % y un rango de 60 a 6,25 %; mientras que al final del tratamiento con ozono disminuyó a una media de 17,5 % y un rango de 16,25 a 18,75 %. En este estudio preliminar se buscó comprobar si existía o no inhibición del porcentaje de agregación plaquetaria después del tratamiento con ozono, igual a lo observado por otros autores in vitro. Se observó una disminución del porcentaje de agregación plaquetaria al final del tratamiento con ozono, con respecto al valor inicial. Los pacientes incluidos en este estudio tuvieron hiporreactividad plaquetaria, pues no se logró alcanzar en la mayoría 50 % de agregación con concentraciones de epinefrina de hasta 10-5 mol/L, que son suficientes para producir este nivel de agregación en individuos sanos. Esto puede deberse a que, al revisar los tratamientos médicos concomitantes se comprobó, que los pacientes consumían fármacos que no son considerados genéricamente antiagregantes plaquetarios, pero que poseen este efecto.5 Entre estos fármacos se encuentran clorpromazina, defenidramina, fenilbutazona, ibuprofen, imipramina, indometacina, naproxeno propanolol, teofilina, trifluorperazina y otros.
A pesar de la hiporreactividad plaquetaria inicial, el tratamiento con ozono estuvo asociado con una reducción adicional en la agregación plaquetaria determinada en el estudio final.
No se estudiaron los parámetros mediadores del estrés oxidativo, pues en este estudio preliminar se buscaba comprobar si existía o no inhibición del porcentaje de agregación plaquetaria después del tratamiento con ozono, igual a lo observado por otros autores in vitro.
Se observó una disminución del porcentaje de agregación plaquetaria al final del tratamiento con ozono, con respecto al valor inicial.
mirian sujeidy marrero 85500
ResponderEliminarDAMEL E. CEDEÑO 85604
ResponderEliminarHEMOSTASIA
• La sangre es un tejido líquido que ejerce múltiples funciones esenciales en
el organismo
• El sistema hemostático es el responsable de mantener a la sangre en
estado líquido, prevenir la pérdida de sangre cuando un vaso se rompe y
evitar la formación de coágulos indeseables
• 2 fases:
– Hemostasia primaria: respuesta del vaso y las plaquetas para formar un tapón
hemostático primario, inestable
– Hemostasia secundaria: formación del coágulo de fibrina. 4 sistemas:
Coagulación
• Anticoagulación
• Fibrinolisis
• Antifibrinolisis
COAGULACIÓN
• Sistema de la coagulación formado por diversas proteínas, casi todas
enzimas (síntesis hepática), que circulan de forma inactiva (proenzimas) y
que se activan unas a otras secuencialmente formando una cascada de
reacciones enzimáticas, tiene lugar sobre una superficie, bien sobre las
plaquetas o sobre la membrana de la pared del vaso y requieren la
presencia de un cofactor no enzimático y de una superficie fosfolipídica
cargada negativamente, que generalmente no está expuesta a la sangre
en condiciones normales, lo que hace que se circunscriba el coágulo.
• Existen dos vías de inicio de la coagulación:
– Vía intrínseca: etapa inicial es la activación del factor XII
– Vía extrínseca: se inicia por la exposición de factor tisular al torrente circulatorio.
El mecanismo en cascada de la coagulación asegura la rápida y explosiva
formación de trombina y del coágulo de fibrina, pero para evitar que todo
el árbol vascular se coagule debe existir un mecanismo de control que
limite el proceso coagulante
• Existen 3 principales mecanismos para este control
– El flujo sanguíneo: dispersa la mayor parte de las enzimas generadas, evitando una
concentración elevada y permitiendo su inhibición por los inhibidores específicos.
– Una superficie cargada negativamente: limita a la zona vascular dañada
– Inhibidores específicos de la coagulación
86070
ResponderEliminarCoagulacion de la sangre:
La habilidad del cuerpo para controlar el flujo de sangre luego de una lesión vascular es un componente indispensable de la supervivencia. El proceso de la coagulación sanguínea y luego la disolución del coágulo, seguido por una reparación del tejido lesionado, se denomina hemostasis. La hemostasis se conforma de 4 eventos principales que ocurren en un orden determinado luego de la pérdida de la integridad vascular:
1. La fase inicial del proceso es la constricción vascular. Esto limita el flujo sanguíneo al área de la lesión.
2. A continuación, se activan las plaquetas por la trombina y se agregan en el sitio de la lesión, formando un tampón temporario y flojo conformado de plaquetas. La proteína fibrinógeno es principalmente responsable de estimular la agregación plaquetaria. Las plaquetas se agregan al unirse al colágeno que se expone debido a la ruptura del recubrimiento epitelial de los vasos. Luego de su activación, las plaquetas liberan ADP un nucleótido y un eicosanoide, TXA2 (los cuales activan más plaquetas), serotonina, fosfolípidos, lipoproteínas y otras proteínas importantes de la cascada de coagulación. Además de su secreción, las plaquetas activadas cambian su conformación para acomodar la formación del coágulo.
3. Para asegurar la estabilidad del tampón flojo inicial, se forma una malla de fibrina (también llamada un coágulo) que recubre al tampón. Si el tampón únicamente contiene plaquetas se denomina un trombo blanco; si glóbulos rojos están presentes se lo denomina un trombo rojo
4. Finalmente, el coágulo debe ser disuelto para que el flujo sanguíneo normal pueda resumir luego de que se repare el tejido. La disolución del coágulo ocurre a través de la acción de la plasmina
Dos vías llevan a la formación de un coágulo de fibrina: la vía intrínseca y la vía extrínseca. Aunque las dos son iniciadas por mecanismos diferentes, las dos convergen en una vía común que lleva a la formación del coágulo. La formación del trombo rojo ó coágulo en respuesta a una anormalidad en un vaso pero en la ausencia de una lesión del tejido es el resultado de la vía intrínseca. La vía intrínseca tiene poca significancia bajo condiciones fisiológicas normales. El suceso más importante clínicamente es la activación de la vía intrínseca por el contacto de la pared del vaso con las partículas de lipoproteína, VLDLs y quilomicrones. Este proceso claramente demuestra el papel de la hiperlipidemia en la generación de la ateroesclerosis. La vía intrínseca también puede ser activada por el contacto de la pared del vaso con bacterias.
La formación del coágulo de fibrina en respuesta a una lesión del tejido es el evento clínico más importante de la hemostasis bajo condiciones normales de respuesta. Este proceso es el resultado de la activación de la vía extrínseca.
Ambas vías son complejas e incluyen varias proteínas diferentes denominadas factores de la coagulación
86070
ResponderEliminarActivación del Fibrinógeno a Fibrina
El fibrinógeno (factor I) consiste en 3 pares de polipéptidos ([Aα][Bβ][γ])2. Las 6 cadenas están covalentemente unidas cerca de sus N-terminales a través de uniones disulfuro. Las porciones A y B de las cadenas Aα y Bβ constituyen los fibrinopéptidos, A y B, respectivamente. Las regiones de fibrinopéptidos del fibrinógeno contienen varios residuos de glutamato y aspartato impartiendo una alta carga negativa a esta región y promoviendo la solubilidad del fibrinógeno en el plasma. La trombina activa es una proteasa de serina que hidroliza al fibrinógeno en cuatro uniones arg-gly (R-G) entre el fibrinopéptido y las porciones a y b de la proteína.
La liberación de los fibrinopéptidos mediada por la trombina, genera monómeros de fibrina con una estructura de subunidades (αβγ)2. Estos monómeros se agregan espontáneamente en un arreglo regular, formando un coágulo de fibrina relativamente débil. Además de la activación de la fibrina, la trombina convierte el factor XIII al factor XIIIa, una transglutaminasa altamente específica que introduce uniones covalentes entre el nitrógeno de amidas de las glutaminas y el grupo ε-amino de las lisinas pertenecientes a los monómeros de fibrina.
Disolución de los Coágulos de Fibrina
La degradación de los coágulos de fibrina es la función de la plasmina, una proteasa de serina que circula como la proenzima inactiva plasminógeno. Cualquier plasmina libre circulante es rápidamente inhibida por la antiplasmina α2. El plasminógeno se une al fibrinógeno y a la fibrina, y así se incorporan en un coágulo. El activador tisular del plasminógeno (tPA) y, a un grado menor, la urocinasa son proteasas de serina que convierten el plasminógeno a plasmina. El tPA inactivo es liberado de las células endoteliales vasculares luego de una lesión; se une a la fibrina y es consecuentemente activado. La urocinasa es producida como el precursor de la prourocinasa por las células epiteliales que recubren los conductos excretorios. El papel de la urocinasa es activar la disolución de los coágulos de fibrina que pueden depositarse en estos conductos.
La tPA activa cliva al plasminógeno para producir plasmina la cual digiere a la fibrina; esta degradación resulta en un producto soluble al cual ni la plasmina ni el plasminógeno se pueden unir. Luego de la liberación del plasminógeno y la plasmina, éstos se inactivan rápidamente por sus respectivos inhibidores. La inhibición de la actividad de la tPA resulta de la unión de específicas proteínas inhibidoras. Por lo menos 4 diferentes inhibidores han sido identificados, dos de los cuales son inhibidores de los activadores del plasminógeno tipo I (PAI-1) y tipo 2 (PAI-2) tienen la mayor importancia fisiológica.
Este comentario ha sido eliminado por el autor.
ResponderEliminar86070:
ResponderEliminarQue son los COAGULOS Y TROMBOS SANGUINEOS?
Los coágulos sanguíneos (coágulos de fibrina) son las masas que se forman cuando la sangre se coagulaLos coágulos sanguíneos o de fibrina son masas que se forman cuando la sangre se coagula y un trombo es un coágulo de sangre que se forma en un vaso o dentro del corazón y permanece allí. Un émbolo es un trombo que viaja desde el vaso o la cámara del corazón donde se formó a otro lugar del cuerpo y el trastorno causado se llama embolia o embolismo (por ejemplo, embolia pulmonar).
Complicaciones
Los trombos y émbolos pueden adherirse fírmemente a un vaso sanguíneo y bloquear parcial o completamente el flujo de sangre en dicho vaso. Esta obstrucción priva a los tejidos en esa área del flujo normal de sangre y oxígeno. Esta condición se llama isquemia y, de no tratarse rápidamente, puede ocasionar daño e incluso la muerte de los tejidos (infarto y necrosis) en dicha área.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarPLAQUETAS, COAGULACION Y FIBRINOLISIS.
Activación plaquetaria.
Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro, derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos.
Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva. Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebro vascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores; cualquier anormalidad o enfermedad de las plaquetas es denominada trombocitopatía, la cual puede ser, ya sea un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Hay desórdenes que pueden reducir el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos, pero en cambio otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragia.
Activación
La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP.
Las células endoteliales producen una proteína llamada factor de von Willebrand (FvW), un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas. Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo.
Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio.
JESUS RANSEL HERNANDEZ VASQUEZ 87063.
ResponderEliminarLa activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria, estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
La agregación plaquetaria, usa el fibrinógeno y el FvW como agentes conectores. El receptor de agregación plaquetaria más abundante es la glicoproteina IIb/IIIa (gpIIb/IIIa); se trata de un receptor para el fibrinógeno dependiente del calcio, fibronectina, vitronectina, trombospondina, y factor de von Willebrand (FvW). Otros receptores incluyen el complejo GPIb-V-IX (FvW) y GPVI (colágeno).o Las plaquetas activadas se adherirán, vía glicoproteína (GP) al colágeno expuesto por el daño epitelial. La agregación y adhesión actúan juntos para formar el tapón plaquetario. Los filamentos de Miosina y actina en las plaquetas son estimuladas para contraerse durante la agregación, reforzando todavía más el tapón.
La agregación plaquetaria es estimulada por el ADP, tromboxano, y la activación del receptor-α2, pero inhibida por agentes antiinflamatorios como las prostaglandinas PGI2 y PGD2. La agregación plaquetaria se ve aumentada por la administración exógena de esteroides anabólicos.
Coagulación sanguínea, cascada de la coagulación.
JESUS RANSEL HERNANDEZ VASQUEZ 87063.
ResponderEliminarCoagulación sanguínea, cascada de la coagulación.
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre. Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse. Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas. Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
Factores de coagulación
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc. En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares. A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola. Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart
JESUS RANSEL HERNANDEZ VASQUEZ 87063.
ResponderEliminarEtapas de la cascada de coagulación, diapositivas 2,3,4,5.
La cascada de coagulación se divide para su estudio, clásicamente en tres vias: La vía intrínseca, la vía extrínseca y la vía común. Las vías intrínseca y extrínseca son las vías de iniciación de la cascada, mientras que la vía común es hacia donde confluyen las otras dos desembocando en la conversión de fibrinógeno en fibrina.
Esta división es un tanto arbitraria y tiene más que ver con las deficiencias de las técnicas que en su momento se utilizaron para desentrañar los mecanismos implicados, que con lo que ocurre realmente en una lesión vascular; ya que en este último caso se establecen varias interrelaciones entre las vías de iniciación.
Mecanismo básico
Cada reacción de estas vías da como resultado el ensamblado de un complejo compuesto por una enzima (factor de coagulación activado), un sustrato (proenzima de un factor de coagulación) y un cofactor que actúa posibilitando la reacción.
Estos componentes se ensamblan en general sobre una superficie fosfolipídica y se mantienen unidos por medio de puentes formados por iones Ca2+. Por lo tanto la reacción en cascada tiende a producirse en un sitio donde este ensamblaje puede ocurrir; por ejemplo sobre la superficie de plaquetas activadas. Tanto la vía intrínseca como la vía extrínseca desembocan en la conversión del factor X en Xa (la letra "a" como subíndice "a" significa "activado") punto en el que se inicia la vía común.
Vía intrínseca
Recibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso hemostático. El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular.
En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación. En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como la celulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción.
A esta vía es posible subdividirla en tres pasos: vía intrínseca.
Formación del factor XIa.
En esta etapa participan cuatro proteínas: Precalicreína, Quininógeno de alto peso molecular (HMWK) y los factores XII y XI. Esta etapa no requiere de iones calcio. Estos cuatro factores se adsorben sobre la superficie cargada negativamente, formando el complejo cebador o de iniciación. De estos factores el XII funciona como verdadero iniciador, ya que si bien es una proenzima, posee una pequeña actividad catalítica que alcanza para activar a la precalicreína convirtiéndola en calicreína.
En segunda instancia la calicreína actúa catalíticamente sobre el factor XII para convertirlo en XIIa, una enzima muchísimo más activa. La actividad catalítica de la calicreína se ve potenciada por el HMWK. Por último la proteasa XIIa actúa sobre el factor XI para liberar XIa.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarFormación del factor IX
El factor IX se encuentra en el plasma como una proenzima. En presencia de iones Ca2+ el factor XIa cataliza la ruptura de una unión peptídica en la molécula del factor IX para formar un glucopéptido de 10 KDa y liberar por otro lado al factor IXa. El factor IX se encuentra ausente en personas con hemofilia tipo B.
Formación del factor Xa
Sobre la membrana de las plaquetas se forma un complejo constituido por los factores IXa, X y VIII, Los residuos gamma-carboxiglutamato de los factores IXa y X actúan como quelantes del ion Ca2+, permitiendo que estos componentes formen un complejo unido por medio de puentes de iones calcio y ayudando a que el complejo se ancle a los fosfolípidos de membrana.
Primero se unen los factores X y IXa a la membrana y luego se une el VIII. El factor VIII es en realidad un homorodímero, formado por cuatro cadenas proteicas, cada una codificada por un gen diferente (VIII:C y VIII:R). El componente VIII: C es conocido como "componente antihemofílico" y actúa como cofactor del IXa en la activación del factor X, el componente VIII: R es el que permite la unión del factor VIII al complejo. La ausencia del componente antihemofílico causa hemofilia A. El complejo formado por los factores IXa-X-VIII-Fosfolípidos y Ca2+ actúa sobre el factor X para convertirlo en Xa. En este punto concluye la vía intrínseca.
Vía extrínseca.
Recibió este nombre debido a que fue posible notar desde un primer momento que la iniciación de esta vía requería de factores ajenos a la sangre. Cuando la sangre entra en contacto con tejidos lesionados o se mezcla con extractos de tejidos, se genera muy rápidamente factor Xa. En este caso la activación de la proenzima X es mediada por un complejo formado por factor VII, Ca2+ y factor tisular unido a fosfolípidos provenientes de las membranas celulares rotas y de las plaquetas (antiguamente este complejo factor tisular-fosfolípidos era conocido como tromboplastina).
El factor tisular es una lipoproteína sintetizada en el endotelio de los vasos sanguíneos de todos los tejidos, aunque es especialmente abundante en pulmón, cerebro y placenta. El factor tisular se encuentra normalmente "secuestrado" en el interior de las células endoteliales y es secretado en respuesta a una lesión, o bajo el efecto de algunas citoquinas tales como el Factor de Necrosis Tumoral (TNF), Interleucina 1 (IL-1); o por endotoxinas bacterianas. La vía extrínseca es muy rápida, se cumple en apenas unos segundos y comprende dos pasos; mientras que la intrínseca insume varios minutos.
Formación del factor VIIa
En primera instancia el factor VII se une a la porción fosfolipídica del factor tisular gracias a sus residuos gamma-carboxiglutamato, utilizando iones Ca2+ como puentes. Este complejo provoca la activación del factor VIIa.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarFormación del factor Xa
El complejo VIIa-III-Ca2+ actúa sobre el factor X convirtiéndolo en la proteasa activa Xa. En este punto termina la vía extrínseca y se inicia la vía común.
Vía común
Llegando al punto en que se activa el factor X, ambas vías confluyen en la llamada vía común. La vía común termina con la conversión de fibrinógeno en fibrina, y el posterior entrecruzamiento de la misma estabilizando el coágulo.
La vía común implica tres etapas:
Formación de trombina
La trombina (también llamada factor II a) es una proteasa generada por la ruptura de la cadena proteica de la proenzima protrombina (factor II), una glicoproteína constituida por 582 aminoácidos y con 12 puentes disulfuro intracatenarios.
La trombina se activa luego de que la proteasa Xa hidroliza dos uniones peptídicas de la protrombina. La Xa produce en primer término la escisión de un fragmento de 32 KDa de la región N-terminal de la cadena, cortándola sobre una unión arginina-treonina. En segundo término produce la ruptura de un enlace entre una arginina y una isoleucina; sin embargo estos dos últimos fragmentos permanecen unidos por un puente disulfuro. La trombina es una serina-proteasa similar a la tripsina, pero mucho más selectiva. Ataca casi de manera exclusiva las uniones arginina con un aminoácido cargado positivamente en sus sustratos.
La conversión de protrombina a trombina debida al factor Xa se acelera notablemente por la formación de un complejo con el factor Va y Ca2+ sobre la superficie de las membranas plaquetarias (fosfolípidos de membrana). El factor Xa y la protrombina se adsorben sobre la membrana utilizando iones Ca2+ como puentes. El factor Va se une a la protrombina acelerando la reacción. El factor Va se produce por la acción de la trombina sobre el factor V en un claro ejemplo de una reacción que va acelerándose a medida que progresa (reacción autoacelerada).
Formación de fibrina
El fibrinógeno (factor I) es una glicoproteína compuesta por seis cadenas polipeptídicas: dos A-alfa, dos B-beta y dos gamma; unidas entre sí por puentes disulfuro. Se trata de una molécula alargada y simétrica formada por tres dominios globulares conectados por segmentos fibrilares.
Cada mitad de la molécula se encuentra formada por tres cadenas (A-alfa, B-beta y gamma) que se enrollan en una triple hélice muy compacta en los sectores fibrilares. Los extremos amino de las seis cadenas se reúnen en el dominio globular central. En un hecho que parecería muy curioso, los extremos N-terminales de las cadenas A-alfa y B-beta emergen como cabos libres del dominio globular central.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarRepresentación de la molécula de fibrinógeno y cómo, al eliminarse los fibrinopéptidos, polimeriza para formar un agregado de fibrina. Estas cadenas son muy ricas en aspartato y glutamato, además las cadenas B-beta poseen en esta región residuos tirosina-O-sulfato formados postraduccionalmente. Estos residuos con una alta tendencia a adquirir carga negativa contribuyen a formar una región central con una muy alta densidad de carga.
Esta región electronegativa central es la responsable de la repulsión entre moléculas de fibrina que las mantiene en solución. La trombina ataca los enlaces arginina-glicina presentes en estos "cabos libres", separando cuatro péptidos; dos segmentos A de 18 aminoácidos cada uno (provenientes de las cadenas A-alfa), y dos segmentos B de 20 aminoácidos (provenientes de las cadenas B-beta). A estos péptidos se los suele denominar "fibrinopéptidos".El resto que queda de la molécula es un monómero de fibrina de composición alfa2beta2gamma.
Al eliminarse los fibinopéptidos desaparecen las fuerzas de repulsión intermoleculares con lo que los monómeros de fibrina tienden a agruparse espontáneamente formando asociaciones altamente ordenadas.
Entrecruzamiento de la fibrina
Los haces paralelos de fibrina polimerizada forman una asociación laxa, que se encuentra en equilibrio con la forma monomérica de la molécula; por lo que sería imposible que cumplieran su papel de formar un coágulo estable sin reforzar esta estructura por medio de enlaces covalentes entre hebras vecinas. La formación de estos "puentes" covalentes intercatenarios es catalizada por la enzima transglutaminasa (conocida también como factor XIIIa). La transglutaminidasa cataliza la formación de enlaces amida entre restos glutamina y lisina de hebras próximas entre sí. En la reacción se libera amoníaco en forma de ion amonio (NH4+). Esta enzima se forma a partir del factor XIII por acción de la trombina.
Regulación y modulación de la cascada (de forma general).
Debido a que la cascada de coagulación consiste en una serie de reacciones que van amplificándose y acelerándose en cada paso, es lógico pensar que debe existir algún mecanismo de regulación; un "freno" a la reacción en cadena; ya que de progresar sin control en pocos minutos podría provocar un taponamiento masivo de los vasos sanguíneos (trombosis diseminada).
Varios mecanismos intervienen en la regulación de la cascada de reacciones:
El flujo sanguíneo normal, arrastra a los factores activados, diluyendo su acción e impidiéndoles acelerarse. Esta es una de las razones por las cuales cuando existe estasis del flujo sanguíneo se favorece la formación de trombos.
El hígado actúa como un filtro quitando de la sangre en circulación los factores activados e inactivándolos.
Existen además algunas proteasas que degradan específicamente a ciertos factores activados, y otras que ejercen acciones inhibitorias sobre factores activos.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarCONTINUACIÓN DE LA CASCADA DE COAGULACION.
Proteína C
La proteína C es una proenzima que se encuentra normalmente en el plasma, y cuya síntesis en el hígado es dependiente de la vitamina K. Esta proteína es convertida en una proteasa activa por la acción de la trombina. La proteína Ca actúa específicamente degradando a los factores Va y VIIIa, con lo que limita la proyección de la cascada. Es interesante notar el triple papel que desempeña la trombina: cataliza la formación de fibrina, activa a la enzima responsable de su entrecruzamiento, y una vez que el proceso de coagulación y estabilización del coágulo está en marcha; ejerce acciones tendientes a limitarlo.
Antitrombina III
La antitrombina III es una glicoproteína de 60 kDa sintetizada en el hígado sin depender de la vitamina K, es considerada la principal inhibidora de la coagulación. Esta proteína actúa inhibiendo irreversiblemente a varios factores procoagulantes activos, el principal de los cuales es la trombina; aunque también actúa sobre la calicreína y los factores IXa, Xa, XIa y XIIa. La acción de la antitrombina es notablemente aumentada por el heteropolisacárido heparina. La heparina se encuentra en el endotelio de los vasos sanguíneos y en los gránulos de las células cebadas, tiene una poderosa acción anticoagulante ya que facilita la unión de la antitrombina III con los factores procoagulantes activos. Existen otras anti-proteasas sanguíneas que también ejercen acción anticoagulante aunque menos potente tales como la alfa2 macroglobulina y la alfa1 antitripsina.
JESUS RANSEL HERNANDEZ VASQUEZ 87063.
ResponderEliminarDIAPOSITIVA 6 TROMBO Y COAGULO.
La trombosis es un coágulo en el interior de un vaso sanguíneo y uno de los causantes de un infarto agudo de miocardio. También se denomina así al propio proceso patológico, en el cual, un agregado de plaquetas o fibrina ocluye un vaso sanguíneo.
Cuando un vaso sanguíneo se lesiona, el cuerpo utiliza plaquetas (trombocitos) y fibrina para formar un coágulo de sangre para prevenir la pérdida de sangre. Incluso cuando un vaso sanguíneo no se lesione, los coágulos de sangre se pueden formar en el cuerpo en ciertas condiciones. Un coágulo que se desprende y comienza a viajar por todo el cuerpo se conoce como embolia.
El trombo es una masa que se forma en el interior del aparato circulatorio y está constituida por la sangre del paciente, específicamente por los elementos sólidos de la sangre. En un individuo vivo la sangre puede coagularse pero esto es fuera del aparato circulatorio por ejemplo, la sangre que pasa al peritoneo, a la pleura o al pericardio. Ahí la sangre coagula y no es un trombo, porque está fuera del aparato cardiovascular.
Las causas son:
Alteración en los vasos sanguíneos:
Arteriosclerosis.
ruptura traumática.
Alteración en los factores de la coagulación:
trombina, protrombina.
disminución de la Proteína C, Proteína S, llamadas estas últimas trombofilias.
Los mecanismos que favorecen la formación de un trombo, son las alteraciones del flujo sanguíneo y estas alteraciones pueden deberse a reposo excesivo en cama (pacientes postoperados). Además en la intervención quirúrgica ha habido una estimulación de los factores de coagulación por la rotura de vasos, la sutura, una serie de intervenciones que involucran al aparato vascular. No es raro que un individuo se opere de una hernia inguinal, y en el momento que se le da de alta y empieza a moverse más de lo que se ha movido en los días anteriores presente una embolia fulminante ocasionándole la muerte. La tercera causa que influye son los componentes de la sangre. Cuando la sangre es más densa disminuyen los líquidos y aumentan los elementos figurados. O hay una hemoconcentración o una policitemia real. Dentro de este se incluye las trombosis a repetición.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarLos sitios de formación de trombo son en el corazón, arterias, venas y capilares, por lo que la trombosis puede formarse en cualquier parte del aparato circulatorio. Otras patologías que pueden provocar una trombosis son aquellas que presentan flujos en torbellinos, como las estrecheces valvulares. Un ejemplo es la estenosis mitral, en donde el flujo en la aurícula se hace más lento y favorece la trombosis. En la estenosis mitral hay que tener en cuenta que lo más probable es que haya trombosis en la orejuela y en alguna parte de la pared de la aurícula. Y si ésa hace flutter o fibrilación, la contracción de la aurícula es ineficiente. Entonces la aurícula no se contrae y además existe oclusión en la salida, de tal modo que hay una lentitud del flujo de salida y por tanto formación de coágulos (trombos).
Otra causa de trombosis es el daño del endotelio. Si un vaso se inflama, por ejemplo una vena de las EEII por un trauma, se produce una lesión de la vecindad y daño endotelial, que desencadena inmediatamente la cascada de coagulación depositándose trombos en la superficie del vaso.
Factores de riesgos
Hay tres tipos de factores de riesgo:
Primarios
Congénitos:
Falta de la proteína C, que evita la coagulación.
Resistencia a la proteína C activada.
Adquiridos:
Anticuerpos antifosfolípido.
Secundarios
Anomalías en las plaquetas.
Anomalías vasculares.
Exceso de tabaco.
Exceso de arsénico (elemento utilizado en el veneno de ratas, por ejemplo).
JESUS RANSEL HERNANDEZ VASQUEZ 87063.
ResponderEliminarTipos de trombosis (trombos).
Las trombosis pueden clasificarse según el nivel de oclusión que alcanzan y el lugar en el que se originan.-Según el grado de oclusión, podemos diferenciar entre trombos ocluyentes y murales. Los primeros son aquellos en los que el vaso queda completamente obstruido por la afección, mientras que en los murales, el resultado es una obstrucción parcial.
Dependiendo de la ubicación, las trombosis se
clasifican según tres tipos: por precipitación, hialina o por coagulación.
Trombosis por precipitación (Trombos Blancos): producida principalmente en arterias o el corazón, se deben al desprendimiento de plaquetas. Son de carácter mural.
Trombosis hialina: producida en vénulas o capilares. Suelen ser provocadas por el desprendimiento de plaquetas y fibrinas.
Trombosis por coagulación (Trombos Rojos): producida en las venas suelen ser de naturaleza oclusiva y deberse a una mezcla de plaquetas y fibrinas, apareciendo estas últimas en una mayor proporción que las primeras.
Esta situación es de extrema gravedad, pues el territorio más allá del trombo deja de recibir irrigación sanguínea, produciéndose inicialmente isquemia y luego muerte de las estructuras. Se puede producir la parálisis de los músculos si se encuentran en el trombo que se ubica en una vena. Dependiendo de la ubicación de la vena, estas trombosis pueden ser graves (trombosis del seno cavernoso), de mediana gravedad (trombosis venosa profunda) o leves (tromboflebitis superficial).
Diferencia entre Trombosis y Embolia
Una trombosis es la obstrucción de un vaso sanguíneo que, generalmente, es producida por una placa de arteriosclerosis que crece en la pared de dicho vaso. Si la placa se desprende, se denomina émbolo y puede ir hacia diversos lugares del organismo, en mayor medida al pulmón. Pero los émbolos pueden ser de material diverso, no solamente trombos, también émbolos infecciosos, de cristales de colesterol, etc. No confundir con la Trombosis hemorroidal.
Tratamiento.
La prevención de coágulos de sangre y el tratamiento reduce los riesgos de ataque al corazón, accidente cerebrovascular y embolia pulmonar. La heparina y warfarina se utilizan a menudo para inhibir la formación y crecimiento de trombos existentes, el primero se une y activa la enzima inhibidor antitrombina III, mientras que el segundo inhibe la vitamina K epóxido reductasa, una enzima necesaria para sintetizar maduros factores de coagulación.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarCoágulos.
Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida.
Causas
Las situaciones en las que es más probable que se forme un coágulo de sangre abarcan:
Estar en reposo en cama por largo tiempo.
Cruzar las piernas durante largos periodos al estar sentado o sentarse durante mucho tiempo, como en un avión o en un vehículo.
Durante y después del embarazo.
No tener suficiente agua en el cuerpo (deshidratación).
Tomar píldoras anticonceptivas u hormonas estrógeno (especialmente en las mujeres que fuman).
Utilizar un catéter intravenoso por largo tiempo.
Los coágulos de sangre también son más probables en personas con cáncer, cirugía o lesión reciente, obesidad y enfermedades del hígado o del riñón. Una acumulación de colesterol que estreche una arteria puede cambiar o reducir el flujo de sangre, facilitando la formación de un coágulo sanguíneo o de un trombo.
Las afecciones que se transmiten de padres a hijos (hereditarias) pueden hacerlo más propenso a la formación de coágulos sanguíneos anormales.
Los trastornos hereditarios que afectan la coagulación son:
Trombofilia por factor V de Leiden
Mutación de la protrombina G20210A
Otras afecciones raras como deficiencias de antitrombina III, proteína C y S
JESUS RANSEL HERNANDEZ VASQUEZ 87063.
ResponderEliminarUn coágulo de sangre puede bloquear una arteria o una vena en el corazón, afectando:
El corazón (angina de pecho o un ataque cardíaco).
Los intestinos (isquemia mesentérica) o trombosis venosa mesentérica.
Los riñones (trombosis de la vena renal).
Las arterias de las piernas o brazos.
Las piernas (trombosis venosa profunda).
Los pulmones (embolia pulmonar).
El cuello o el cerebro (accidente cerebrovascular).
Posibles complicaciones
Los trombos y émbolos pueden fijarse firmemente a un vaso sanguíneo y pueden bloquear parcial o completamente el flujo de sangre a dicho vaso.Un bloqueo en el vaso sanguíneo impide que el flujo normal de sangre y oxígeno llegue a los tejidos en ese sitio, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede ocasionar daño tisular o necrosis en esa área.
JESUS RANSEL HERNANDEZ VASQUEZ 87063.
ResponderEliminarDIAPOSITIVA 7 FIBRINOLISIS.
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos.
Bioquímica
La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinólisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón.
La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca:
En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA.
En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno.
Regulación
La fibrinólisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Consecuencias
Cuando se produce demasiado rápidamente, puede provocar hemorragias espectaculares (fibrinólisis hemorrágica) intra o postoperatorias (particularmente en Cirugía Torácica), o después de un parto, de un aborto o de un choque traumático; o bien puede provocar hemorragias menos importantes (hematuria) en el curso de ciertos cánceres (próstata, páncreas, estómago), de las cirrosis y de las leucosis. Obedece a la liberación de activadores de la profibrinolisina, o bien a la producción de un fermento proteolítico por los tejidos enfermos.
Tipos
Puede ser primitiva aunque por lo general es secundaria, reactiva, en el curso de un síndrome de coagulación intravascular diseminada
Causas, incidencia y factores de riesgo
Los coágulos de sangre se forman a partir de una proteína llamada fibrina. La descomposición de la fibrina (fibrinólisis) puede incrementarse bajo ciertas condiciones, tales como:
Infecciones bacterianas
Ejercicio intenso
Glucemia baja
Oxigenación insuficiente a los tejidos
Procedimiento médico alternativo
En algunas situaciones, es posible que los médicos quieran acelerar el proceso de fibrinólisis. Por ejemplo, cuando se forma un coágulo sanguíneo anormal en los vasos sanguíneos del corazón y provoca un ataque cardíaco, se pueden suministrar sustancias fibrinolíticas sintéticas (como, estreptocinasa o Retavase) para disolverlo.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarDIAPOSITIVA 8 SISTEMA FIBRINOLITICO.
El sistema fibrinolítico va a jugar además un papel importante en diversas situaciones en las que se produce proteolisis tisular, tales como inflamación, invasión tumoral o neovascularización. La importancia fisiopatológica del sistema fibrinolítico deriva de que las alteraciones que condicionan un defecto de actividad fibrinolíticas pueden predisponer a la trombosis, mientras que un exceso de activación favorecería la aparición de hemorragia. Además va a tener una gran importancia en el área terapéutica por la aplicación creciente de fármacos fibrinolíticos en el tratamiento de la trombosis, especialmente en el infarto agudo de miocardio.
Principales componentes del sistema fibrinolítico: La fibrinólisis es un encadenamiento (proceso) enzimático compuesto por una serie de activadores e inhibidores los cuales regulan la conversión del proenzima circulante, plasminógeno, en el enzima activo plasmina.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarLa producción de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina y esto es muy importante para el mantenimiento de la permeabilidad vascular. Los principales componentes del sistema fibrinolítico son los siguientes:
PLASMINÓGENO
Es una glicoproteína monocatenaria compuesta por 790 aminoácidos y 24 puentes disulfuro con un contenido en carbohidratos del 2% y un peso molecular (Pm) de 92.000 daltons, sintetizándose en el hígado; su concentración plasmática es de 2mM (20 mg/dl) y su vida media de 2,2 días. El gen que codifica esta proteína está localizado en el cromosoma 6. En su forma nativa el plasminógeno contiene ácido glutámico en posición amino-terminal (Glu-plasminógeno), pudiendo ser convertido por pequeñas cantidades de plasmina a formas con lisina, valina o metionina en dicha posición, denominadas Lis-plasminógeno, los cuales poseen mayor afinidad por la fibrina y se activan más fácilmente por los activadores fibrinolíticos.
ACTIVADORES DEL PLASMINÓGENO
La activación del plasminógeno a plasmina tiene lugar por escisión del enlace Arg561-Val562 en la molécula del Glu-plasminógeno por acción de los diferentes activadores. La plasmina así formada es una serín-proteasa bicatenaria compuesta por una cadena pesada derivada de la porción amino-terminal, que contiene 560 aminoácidos y 5 “kringles” en donde se localizan los LBS, y una cadena ligera derivada de la posición carboxiterminal compuesta por 241 aminoácidos, que contiene el centro activo formado por los aminoácidos serina, histidina y ácido aspártico. En la actualidad se distinguen varias vías de activación.
Activación intrínseca del plasminógeno
Fue denominada inicialmente activación por contacto o XII-dependiente por estar implicados diversos factores del sistema de contacto de la coagulación. Actualmente se sabe que además del factor XII existen otras sustancias en la sangre, denominadas precursores, como la precalicreína o el quininógeno de alto peso molecular que pueden inducir la activación del plasminógeno. La activación del factor XII conduce a la generación de calicreína que va a ser un potente activador de la fibrinólisis intrínseca.
Activador tisular del plasminógeno (t-PA)
Es una proteasa serínica monocatenaria constituida por 530 aminoácidos y un Pm de 68.000 daltons la cual puede ser convertida en una molécula de doble cadena por acción de la plasmina. La cadena pesada A (1-278aa) contiene dominios homólogos a los encontrados en otras proteínas: una región homóloga a la de la fibronectina, denominada dominio “finger”, una estructura similar al factor de crecimiento epidérmico y dos “kringles” similares a los del plasminógeno.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarCONTINUACION DEL SISTEMA FIBRINOLITICO.
Activadores tipo uroquinasa y prouroquinasa.
La UK es una proteasa serínica similar a la tripsina, compuesta por dos cadenas polipeptídicas de 20.000 y 34.000 daltons unidas por un puente disulfuro; fue aislada inicialmente a partir de orina humana y cultivo de células embrionarias de riñón humano. Es un activador directo del plasminógeno mediante escisión de un enlace Arg561-Val562 con formación de Glu-plasmina pero, a diferencia del t-PA, carece de afinidad específica por la fibrina, ya que activa indiscriminadamente tanto al plasminógeno circulante como al unido a la fibrina. Tanto su mecanismo de acción como sus características farmacocinéticas serán revisadas en profundidad más adelante.
La prouroquinasa es una glicoproteína monocatenaria compuesta por 411 aminoácidos y un Pm de 54.000 daltons. La porción amino-terminal contiene una región homóloga al factor de crecimiento epidérmico y otra similar a los “kringles” del plasminógeno, pero a diferencia del t-PA no posee un dominio “finger, lo que quizás pudiera explicar su falta de afinidad por la fibrina. En la región carboxi-terminal se encuentra su centro activo el cual contiene el aminoácido serina.
Activación exógena del plasminógeno.
Consiste en la activación que se produce por la acción de diferentes sustancias exógenas administradas con fines terapéuticos.
INHIBIDORES DEL SISTEMA FIBRINOLÍTICO
Se clasifican en inhibidores competitivos del plasminógeno, inhibidores de los activadores del plasminógeno e inhibidores de la plasmina.
MODULADORES DE LA ACTIVIDAD FIBRINOLÍTICA.
La actividad fibrinolítica plasmática viene determinada por un balance entre los activadores e inhibidores, fundamentalmente t-PA y PAI-1. Ambas sustancias están sometidas a una estrecha regulación por la acción de diversos estímulos fisiológicos y patológicos. Así, por ejemplo, el estrés, ejercicio físico, oclusión venosa o administración de fármacos vasoactivos aumentan la actividad fibrinolítica plasmática al favorecer la liberación de t-PA desde el endotelio vascular.
La insulina aumenta la síntesis de PAI-1 por los hepatocitos, aunque no tiene efecto sobre las células endoteliales. Los corticoides y especialmente la dexametasona, disminuyen la liberación de t-PA e inducen la síntesis de PAI-1. Diversas citoquinas como la interleuquina 1 (IL-1) y el factor de necrosis tumoral (TNF) inducen una disminución de t-PA y un aumento de PAI-1. Finalmente, la endotoxina, ya sea directamente o a través de citoquinas, inducen un marcado aumento del PAI-1 por el endotelio vascular.
JESUS RANSEL HERNANDEZ VASQUEZ 87063.
ResponderEliminarAPUNTES CLAVES SOBRE LAS PLAQUETAS, DIAPSITIVA 1.
Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro, derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos.
Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva. Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebro vascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores. Cualquier anormalidad o enfermedad de las plaquetas se denomina trombocitopatía,2 la cual puede consistir, ya sea en tener un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Se pueden producir desórdenes que reducen el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos. Sin embargo, otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragias.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarLas plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular; Estos dos factores de crecimiento han demostrado jugar un papel significativo en la regeneración y reparación del tejido conectivo; Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarCinética
Las plaquetas son producidas en el proceso de formación de las células sanguíneas llamado (trombopoyesis) en la médula ósea, por fragmentación en los bordes citoplasmáticos de los megacariocitos. El rango fisiológico de las plaquetas es de 150-400 x 109/litro. Un adulto sano produce cada día alrededor de 1 x 1011 plaquetas de media. La expectativa de vida de las plaquetas circulantes es de 7 a 10 días. La producción de megacariocitos y plaquetas está regulada por la trombopoyetina, una hormona producida habitualmente por el hígado y los riñones. Cada megacariocito produce entre 5.000 y 10.000 plaquetas, Las plaquetas son destruidas por fagocitosis en el bazo y por las células de Kupffer en el hígado.
Una reserva de plaquetas es almacenada en el bazo y son liberadas cuando se necesitan por medio de contracción esplénica mediada por el sistema nervioso simpático.
Formación de trombos.
La función plaquetaria consiste en el mantenimiento del corazón; Esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio.
Activación.
La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP. Las células endoteliales producen una proteína llamada factor de von Willebrand (FvW),un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas.
Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo.
Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio.
La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria (plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
Cambio de forma.
Las plaquetas activadas cambian su forma haciéndose más esféricas, y formando pseudopodos en su superficie. De esta forma toman una forma estrellada.
JESUS RANSEL HERNNADEZ VASQUEZ 87063
ResponderEliminarSecreción de gránulos.
Las plaquetas contienen gránulos alfa y gránulos densos. Las plaquetas activadas excretan el contenido de estos gránulos dentro de sus sistemas canaliculares y en la sangre circundante. Existen dos tipos de gránulos:
Gránulos densos (contienen ADP o ATP, calcio, y serotonina).
Granulos-α (contienen factor 4 plaquetario, factor de crecimiento transformante beta 1 (TGF beta 1), factor de crecimiento derivado de plaquetas, fibronectina, B-tromboglobulina, FvW, fibrinógeno, y factores de coagulación factor V y XIII).
Síntesis de tromboxano A2.
La activación plaquetaria inicia la vía del ácido araquidónico para producir Tromboxano A2; el tromboxano A2 está involucrado en la activación de otras plaquetas y su formación es inhibida por los inhibidores de la COX, como el ácido acetilsalicílico.
Adhesión y agregación.
La agregación plaquetaria, usa el fibrinógeno y el FvW como agentes conectores. El receptor de agregación plaquetaria más abundante es la glicoproteina IIb/IIIa (gpIIb/IIIa); se trata de un receptor para el fibrinógeno dependiente del calcio, fibronectina, vitronectina, trombospondina, y factor de von Willebrand (FvW). Otros receptores incluyen el complejo GPIb-V-IX (FvW) y GPVI (colágeno).ó Las plaquetas activadas se adherirán, via glicoproteína (GP) Ia, al colágeno expuesto por el daño epitelial. La agregación y adhesión actúan juntos para formar el tapón plaquetario. Los filamentos de Miosina y actina en las plaquetas son estimuladas para contraerse durante la agregación, reforzando todavía más el tapón.
La agregación plaquetaria es estimulada por el ADP, tromboxano, y la activación del receptor-α2, pero inhibida por agentes antiinflamatorios como las prostaglandinas PGI2 y PGD2. La agregación plaquetaria se ve aumentada por la administración exógena de esteroídes anabólicos.
Reparación de heridas.
El coágulo sanguíneo es solo una solución temporal para detener la hemorragia; la reparación del vaso debe ocurrir después. La agregación plaquetaria ayuda en este proceso mediante la secreción de sustancias químicas que promueven la invasión de fibroblastos del tejido conectivo adyacente hacia el interior de la herida para formar una costra. El coágulo obturador es lentamente disuelto por la enzima fibrinolítica, plasmina, y las plaquetas son eliminadas por fagocitosis.
Otras funciones
Retracción del coágulo
Pro-coagulación
Inflamación
Señalización citoquínica
Fagocitosis
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarSeñalización citoquínica
Adicionalmente a su función de ser el efector celular de la hemostasia, las plaquetas son rápidamente depositadas en sitios de lesión o infección, y potencialmente modulan los procesos inflamatorios por medio de la interacción con leucocitos y por la secreción de citoquinas, quimiosinas, y otros mediadores de la inflamación las plaquetas también secretan factor de crecimiento derivado de plaquetas (PDGF).
Papel en enfermedades.
Recuentos altos y bajos.
El recuento de plaquetas de un individuo sano se encuentra entre 150,000 y 450,000 por μl (microlitro) de sangre (150-450 x 109/L).18 El 95 % de los individuos sanos tendrán recuentos de plaquetas dentro de este rango. Algunos tendrán recuentos de plaquetas estadísticamente anormales sin tener ninguna anormalidad demostrable. Sin embargo, si el recuento es muy alto o muy bajo la probabilidad de que una anormalidad este presente es más alta.
Tanto la trombocitopenia como la trombocitosis pueden manifestarse como problemas de coagulación. En general, los recuentos bajos de plaquetas incrementan el riesgo de sangrado; sin embargo existen excepciones. Por ejemplo la trombocitopenia inmune inducida por heparina. En la trombocitosis se puede producir trombosis, sin embargo esto sucede principalmente cuando el recuento elevado es debido a desordenes mieloproliferativos.
Los recuentos de plaquetas en general, no son corregidos con transfusión a menos que el paciente esté sangrando o el recuento haya caído por debajo 5 x 109/L. La transfusión está contraindicada en la púrpura trombocitopénica idiopática (PTI), puesto que estimula la coagulopatía. En los pacientes sometidos a cirugía, niveles inferiores a 50 x 109/L están asociados a sangrado quirúrgico anormal, y procedimientos anestésicos regionales como la anestesia epidural son evitados para niveles inferiores a 80-100 x 109/L.
El recuento normal de plaquetas no es garantía de función adecuada. En algunos estados, las plaquetas, siendo normales en número, son disfuncionales. Por ejemplo, el ácido acetilsalicílico interrumpe irreversiblemente la función plaquetaria mediante la inhibición de la ciclooxigenasa-1 (COX1), y por consiguiente la hemostasia normal. Las plaquetas resultantes no tienen ADN y son incapaces de producir nueva ciclooxigenasa. La función plaquetaria normal no se restaurara hasta que el uso de ASA haya cesado y un número suficiente de las plaquetas afectadas hayan sido reemplazadas por nuevas, lo cual suele tardar unos 7 días. El Ibuprofeno, un AINE, no tiene un período tan largo de efecto, y la función plaquetaria vuelve a la normalidad dentro de las 24 horas, y tomando ibuprofeno antes que el ASA prevendrá los efectos irreversibles de esta. La Uremia, a consecuencia de la insuficiencia renal, conduce a la disfunción plaquetaria que puede ser aminorada con la administración de desmopresina.
JESUS RANSEL HERNANDEZ VASQUEZ 87063
ResponderEliminarMedicamentos
• Agentes orales usados a menudo para alterar/suprimir la función plaquetaria:
• Ácido acetilsalicílico
• Clopidogrel
• Cilostazol
• Ticlopidina
Agentes intravenosos usados a menudo para alterar/suprimir la función plaquetaria:
• Abciximab
• Eptifibatida
• Tirofiban
Enfermedades.
Desordenes que provocan un recuento bajo de plaquetas:
• Trombocitopenia
• Púrpura trombocitopénica idiopática
• Púrpura trombocitopénica trombótica
• Púrpura trombocitopenica inducida por medicamentos
• Enfermedad de Gaucher
• Anemia aplásica
• Trastornos Aloimunes
• Trombocitopenia fetomaterna autoinmune
• Algunas reacciones trasfusionales
Desordenes que provocan disfunción o recuento reducido:
• Síndrome HELLP
• Síndrome urémico hemolítico
• Quimioterapia
• Dengue
• Deficiencia del almacenamiento en gránulos Alfa-Delta; es un desorden hemorrágico hereditario.
Desordenes caracterizados por recuentos elevados:
• Trombocitosis, incluyendo trombocitosis esencial (recuento elevado, ya sea reactivo o como una expresión de trastorno mieloproliferativo); puede mostrar plaquetas disfuncionales.
• Desordenes de la agregación y adherencia plaquetarios:
• Síndrome de Bernard-Soulier
• Tromboastenia de Glanzmann
• Síndrome de Scott's
• Enfermedad de von Willebrand
• Síndrome de Hermansky-Pudlak
• Síndrome de plaquetas grises
• Desordenes del metabolismo de plaquetas
• Actividad disminuida de la oxigenación, inducida o congénita
• Defectos del almacenamiento, adquirido o congénito
Desordenes que comprometen la función plaquetaria:
• Hemofilia
• Desordenes en los cuales las plaquetas juegan un papel clave:
• Aterosclerosis
• Enfermedad coronaria arterial, e infarto del miocardio
• Accidente cerebro vascular
• Enfermedad arterial oclusiva periférica
• Cancer
JESUS RANSEL HERNANDEZ VASQUEZ 87063.
ResponderEliminarPlasma rico en plaquetas (PRP).
La preparación del plasma rico en plaquetas (PRP) de procedencia autóloga, requiere la extracción y recolección de sangre periférica del paciente, la separación de las plaquetas y el plasma de los otros elementos formes sanguíneos, y la posterior polimerización de la fibrina de dicho plasma para concentrar las plaquetas formando un gel rico en plaquetas con suficiente estabilidad como para ser implantado quirúrgicamente. Actualmente, algunos métodos comerciales para la preparación del PRP utilizan calcio y trombina bovina o bien, trombina preparada de forma autóloga para crear una matriz rica en plaquetas y fibrina. La preparación de trombina autóloga requiere tiempo y pasos adicionales, así como un mayor volumen de sangre; por otro lado, el uso de trombina bovina ha sido asociado con el desarrollo de anticuerpos contra los factores de coagulación V y XI, y la misma trombina, aumentando de esta forma el riesgo de anormalidades en la coagulación. Adicionalmente, para asegurar una degranulación de las plaquetas y la formación de un coágulo estable, se utilizan grandes cantidades de trombina, esto puede causar una liberación inmediata de los factores de crecimiento.
La liberación de factores de crecimiento es desencadenada por la activación de las plaquetas, esta puede ser iniciada por una gran variedad de sustancias o estímulos como la trombina, el cloruro de calcio, el colágeno o el adenosina 5c-difosfato. Un coágulo sanguíneo de PRP contiene aproximadamente un 4% de glóbulos rojos, 95% de plaquetas y 1% de glóbulos blancos. Las propiedades del PRP están basadas en los múltiples factores de crecimiento y de diferenciación producidos y liberados a raíz de la activación de las plaquetas. Estos factores son críticos para la regulación y estimulación del proceso curativo de lesiones. Por otro lado, existe una segunda generación del concentrado de plaquetas que recibe el nombre de matriz rica en plaquetas y fibrina la cual es un mejoramiento del PRP preparado tradicionalmente.
Existe un método actual que evita el uso de trombina como activador Este sistema utiliza únicamente calcio y centrifugación para activar la polimerización de la fibrina y formar así el PRFM. PRFM, en forma de gel o una membrana densa y flexible, puede ser aplicada al paciente y la liberación de los factores de crecimiento es desencadenada por los activadores autólogos presentes en el sitio de aplicación. Este método permite una liberación gradual de los factores de crecimiento en el sitio de aplicación, que pueden emitir señales a diferentes tipos celulares para que emitan una respuesta en momentos apropiados. Estudios in vitro indican que el PRFM presenta una liberación gradual y estable de los factores de crecimiento a lo largo de 7 días.
El plasma rico en plaquetas (PRP) puede obtenerse por medio de diferentes técnicas ya sean separadores celulares de propósitos generales o bien, separadores celulares para la concentración de plaquetas. Muchos productos comerciales se encuentran disponibles en este campo, la mayoría de ellos obtienen resultados similares, cuyas diferencias se deben fundamentalmente al precio, tiempo, espacio requerido y la tecnología necesaria para fabricarlo. Son pocos los productos comerciales disponibles para la obtención de una matriz rica en plaquetas y fibrina como producto final.
stephanie de los santos 86182
ResponderEliminarPlaquetas
Las plaquetas son pequeñas células que circulan en la sangre; participan en la formación de coágulos sanguíneos y en la reparación de vasos sanguíneos dañados.
Cuando un vaso sanguíneo se lesiona, las plaquetas se adhieren al área dañada y se distribuyen a lo largo de la superficie para detener la hemorragia (este proceso se conoce como adhesión).
Los procesos de activación plaquetaria comprenden complejas rutas de señalización que involucran sistemas de proteínas G acoplados a enzimas efectoras como la fosfolipasa A2, fosfolipasa C, adenilato y guanilato ciclasas. Las propiedades funcionales y estructurales de las plaquetas están estrechamente relacionadas, lo que les confiere un alto potencial trombogénico, contribuyendo sustancialmente en el desarrollo de un infarto cerebral.
Coagulación 1: son un grupo de afecciones en las cuales hay un problema con el proceso de coagulación sanguínea del cuerpo. esto puede llevar a que se presente un sangrado intenso y prolongado.
PROBLEMAS DE COAGULACIÓN - 2 -
Deficiencia del factor X
Es un trastorno causado por la presencia de muy poca cantidad de una proteína llamada factor X en la sangre, lo cual lleva a problemas de coagulación.
Causas
Cuando uno sangra, el cuerpo inicia una serie de actividades que ayudan a que la sangre se coagule, lo cual se denomina cascada de la coagulación. El proceso involucra proteínas especiales, llamadas factores de coagulación. Cuando falta uno o más de estos factores de coagulación, generalmente hay una posibilidad más alta de sangrado
Trombo: es una masa intravascular o intracardíaca formada por elementos de la sangre e insolubles en ella, se caracterizan por estar fijos a la pared del vaso por una zona donde son más rígidos y consistentes. Pueden ser arteriales o venosos.
Coagulos: intravasculares son postmorten son gelatinosos, con una porción declive rojo oscura porque los eritrocitos sedimentan por acción de la graverdad y un sobrenadante amarillo en grasa de pollo, no estan fijos a la pared del vaso.
En cambio los trombos son mas consistentes, estan anclados en un punto a la pared del vaso o cardiaca, los arteriales consistentes de color blanco grisaceo y friables, formados por una red de plaquetas fibrina y leucocitos en degeneración.
Fibrinólisis. (De fibrina, y del griego lyein, disolver). La fibrinólisis primaria se refiere a la descomposición normal de los coágulos, mientras que la fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa.
El sistema fibrinolítico va a jugar además un papel importante en diversas situaciones en las que se produce proteolisis tisular, tales como inflamación, invasión tumoral o neovascularización. La importancia fisiopatológica del sistema fibrinolítico deriva de que las alteraciones que condicionan un defecto de actividad fibrinolítica pueden predisponer a la trombosis, mientras que un exceso de activación favorecería la aparición de hemorragia.
karelyn meralis pensa sosa 86961
ResponderEliminarLas plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro,1 derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos.
Si el número de plaquetas es demasiado bajo, puede ocasionar una hemorragia excesiva. Por otra parte si el número de plaquetas es demasiado alto, pueden formarse coágulos sanguíneos y ocasionar trombosis, los cuales pueden obstruir los vasos sanguíneos y ocasionar un accidente cerebro vascular, infarto agudo de miocardio, embolismo pulmonar y el bloqueo de vasos sanguíneos en cualquier otra parte del cuerpo, como en las extremidades superiores e inferiores. Cualquier anormalidad o enfermedad de las plaquetas se denomina trombocitopatía,2 la cual puede consistir, ya sea en tener un número reducido de plaquetas (trombocitopenia), un déficit en la función (tromboastenia), o un incremento en el número (trombocitosis). Se pueden producir desórdenes que reducen el número de plaquetas, como la púrpura trombocitopénica idiopática (PTI) y causan problemas hemorrágicos. Sin embargo, otros como la trombocitopenia inducida por la heparina pueden causar trombosis, o coágulos, en lugar de hemorragias.
Las plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular; Estos dos factores de crecimiento han demostrado jugar un papel significativo en la regeneración y reparación del tejido conectivo; Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.
karelyn meralis pena sosa 86961
ResponderEliminarcoagulacion :
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse.
Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas.
Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo
karelyn meralis pensa sosa 86961
ResponderEliminarLa trombosis es un coágulo en el interior de un vaso sanguíneo y uno de los causantes de un infarto agudo de miocardio. También se denomina así al propio proceso patológico, en el cual, un agregado de plaquetas o fibrina ocluye un vaso sanguíneo.
Cuando un vaso sanguíneo se lesiona, el cuerpo utiliza plaquetas (trombocitos) y fibrina para formar un coágulo de sangre para prevenir la pérdida de sangre. Incluso cuando un vaso sanguíneo no se lesione, los coágulos de sangre se pueden formar en el cuerpo en ciertas condiciones. Un coágulo que se desprende y comienza a viajar por todo el cuerpo se conoce como embolia.1 2
El trombo es una masa que se forma en el interior del aparato circulatorio y está constituida por la sangre del paciente, específicamente por los elementos sólidos de la sangre. En un individuo vivo la sangre puede coagularse pero esto es fuera del aparato circulatorio por ejemplo, la sangre que pasa al peritoneo, a la pleura o al pericardio. Ahí la sangre coagula y no es un trombo, porque está fuera del aparato cardiovascular.
Las causas son:
alteración en los vasos sanguíneos;
arteriosclerosis
ruptura traumática
alteración en los factores de la coagulación;
trombina, protrombina
disminución de la Proteína C, Proteína S, llamadas estas últimas trombofilias.
Los mecanismos que favorecen la formación de un trombo, son las alteraciones del flujo sanguíneo y estas alteraciones pueden deberse a reposo excesivo en cama (pacientes postoperados). Además en la intervención quirúrgica ha habido una estimulación de los factores de coagulación por la rotura de vasos, la sutura, una serie de intervenciones que involucran al aparato vascular. No es raro que un individuo se opere de una hernia inguinal, y en el momento que se le da de alta y empieza a moverse más de lo que se ha movido en los días anteriores presente una embolia fulminante ocasionándole la muerte.
La tercera causa que influye son los componentes de la sangre. Cuando la sangre es más densa disminuyen los líquidos y aumentan los elementos figurados. O hay una hemoconcentración o una policitemia real. Dentro de este se incluye las trombosis a repetición.
Los sitios de formación de trombo son en el corazón, arterias, venas y capilares, por lo que la trombosis puede formarse en cualquier parte del aparato circulatorio.
Otras patologías que pueden provocar una trombosis son aquellas que presentan flujos en torbellinos, como las estrecheces valvulares. Un ejemplo es la estenosis mitral, en donde el flujo en la aurícula se hace más lento y favorece la trombosis. En la estenosis mitral hay que tener en cuenta que lo más probable es que haya trombosis en la orejuela y en alguna parte de la pared de la aurícula. Y si ésa hace flutter o fibrilación, la contracción de la aurícula es ineficiente. Entonces la aurícula no se contrae y además existe oclusión en la salida, de tal modo que hay una lentitud del flujo de salida y por tanto formación de coágulos (trombos).
Otra causa de trombosis es el daño del endotelio. Si un vaso se inflama, por ejemplo una vena de las EEII por un trauma, se produce una lesión de la vecindad y daño endotelial, que desencadena inmediatamente la cascada de coagulación depositándose trombos en la superficie del vaso.
En cuanto a los líquidos extraños que entran al aparato circulatorio y pueden provocar una embolia, estos pueden ser fundamentalmente el líquido amniótico que ingresa a la circulación materna al producirse desprendimiento de la placenta, y rotura de las venas uterinas y/o del cervix constituyendo en una embolia amniótica, ésta dependiendo de la cuantía puede ser fatal.
También en las fracturas múltiples, la medula ósea adiposa de los huesos que es semilíquida puede entrar a la circulación y embolizar hacia el pulmón o cerebro.
Jose Fuentes
ResponderEliminar86966
Activacion Plaquetaria
La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP.
Las células endoteliales producen una proteína llamada factor de von Willebrand,un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el factor de von Willebrand es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas.
Cuando la capa endotelial es lesionada, el colágeno, el factor de von Willebrand y el factor tisular del endotelio son expuestos al flujo sanguíneo.
Cuando las plaquetas hacen contacto con el colágeno o el factor de von Willebrand, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio.
La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria(plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
Coagulación
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
El sistema plasmático de la coagulación, que está compuesto por una serie de factores:
• Factor I o fibrinógeno
• Factor II o protrombina
• Factor III o tromboplastina tisular
• Factor IV, que es el Ca++
• Factor V o proacelerina
• Factor VII o proconvertina
• Factor VIII o antihemofílico A
• Factor IX o Christmas o factor antihemofílico B
• Factor X o de Stuart- Prower
• Factor XI o antecedente de la tromboplastina del plasma Factor XII o de Hageman
• Factor XIII o estabilizador de la fibrina
• Activador de la protrombina o tromboplastina completa Factor de Fitzgerald o kininógeno de alto peso molecular
Fibrinolisis
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro,1 derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos.
ResponderEliminarCuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Victor Rodriguez 88158
EliminarNestor Maione Aponte 2012-0718
ResponderEliminarLas plaquetas son producidas en el proceso de formación de las células sanguíneas llamado (trombopoyesis) en la médula ósea, por fragmentación en los bordes citoplasmáticos de los megacariocitos.
El rango fisiológico de las plaquetas es de 150-400 x 109/litro.
Alrededor de 1 x 1011 plaquetas de media son producidas cada día por un adulto sano.
La expectativa de vida de las plaquetas circulantes es de 7 a 10 días.
La producción de megacariocitos y plaquetas está regulada por la trombopoyetina, una hormona producida usualmente por el hígado y los riñones.
Cada megacariocito produce entre 5.000 y 10.000 plaquetas.
Las plaquetas son destruidas por fagocitosis en el bazo y por las células de Kupffer en el hígado.
Una reserva de plaquetas es almacenada en el bazo y son liberadas cuando se necesitan por medio de contracción esplénica mediada por el sistema nervioso simpático.
Formación de trombos
La función plaquetaria consiste en el mantenimiento de la hemostasia; Esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio.
Activación
La superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP. Las células endoteliles producen una proteína llamada factor de von Willebrand (FvW),un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas. Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo. Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio. La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria; estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
RUENDY PICA 83565
ResponderEliminarACTIVACION PLAQUETARIA:
Un enorme número de sustancias puede actuar sobre las plaquetas e inducir su activación [el factor de activación plaquetaria,6 trombina, el tromboxano (TXA2), fosfato de adenosina (ADP),10 epinefrina, vasopresina y serotonina] a través de sus respectivos receptores, pero todas lo hacen a merced de rutas de señalización intracelular que involucran un sistema de proteínas G acoplado a enzimas efectoras comolas fofolipasas A2 (PLA2) y C (PLC) y adenilato y guanilato ciclasas.5, 6, 10
Hasta ahora se han identificado nueve componentes diferentes de la familia de proteínas G en las plaquetas y, dependiendo del tipo de proteína G que participe en la señalización. Se puede activar o inhibir el efecto.6 Un ejemplo lo constituye la regulación por proteína G de la adenilato ciclasa y por tanto de la producción de monofosfato de adenosina cíclico (AMPc). Un incremento de las cantidades de AMPc bloquea la liberación de calcio en el STD e inhibe la función de la plaqueta. Este es el mecanismo que utilizan las prostaciclinas del endotelio vascular sano para inhibir la activación plaquetaria.
RUENDY PICA 83565
ResponderEliminarCOAGULACION DE LA SANGRE:
Es el proceso por el cual se forma un coágulo sanguíneo. Comienza en respuesta a una lesión en un vaso sanguíneo.
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso, por el cual, la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoléculares en forma de una red tridimensional.
El fibrinógeno, una vez transformado, recibe el nombre de fibrina. Coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse.
Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas.
Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc.
En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares.
A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (precalicreína —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para su síntesis en el hígado, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
REUNDY PICA 83565
ResponderEliminarTROMBO Y COAGULO :
Los coágulos sanguíneos (coágulos de fibrina) son las masas que se forman cuando la sangre se coagula.
Los coágulos sanguíneos o de fibrina son masas que se forman cuando la sangre se coagula y un trombo es un coágulo de sangre que se forma en un vaso o dentro del corazón y permanece allí. Un émbolo es un trombo que viaja desde el vaso o la cámara del corazón donde se formó a otro lugar del cuerpo y el trastorno causado se llama embolia o embolismo (por ejemplo, embolia pulmonar).
Los trombos y émbolos pueden adherirse fírmemente a un vaso sanguíneo y bloquear parcial o completamente el flujo de sangre en dicho vaso. Esta obstrucción priva a los tejidos en esa área del flujo normal de sangre y oxígeno. Esta condición se llama isquemia y, de no tratarse rápidamente, puede ocasionar daño e incluso la muerte de los tejidos (infarto y necrosis) en dicha área.
RUENDY PICA 83565
ResponderEliminarFIBRINOLISIS:
La fibrinólisis primaria se refiere a la descomposición normal de los coágulos, mientras que la fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa.
Cuando se produce demasiado rápidamente, puede provocar hemorragias espectaculares (fibrinólisis hemorrágica) intra o postoperatorias (particularmente en Cirugía Torácica), o después de un parto, de un aborto o de un choque traumático; o bien puede provocar hemorragias menos importantes (hematuria) en el curso de ciertos cánceres (próstata, páncreas, estómago), de las cirrosis y de las leucosis. Obedece a la liberación de activadores de la profibrinolisina, o bien a la producción de un fermento proteolítico por los tejidos enfermos.
Puede ser primitiva aunque por lo general es secundaria, reactiva, en el curso de un síndrome de coagulación intravascular diseminada.
SISTEMA FIBRINOLITICO:
El sistema fibrinolítico endógeno se activa por la presencia de un trombo intravascular, convirtiendo el plasminógeno en plasmina, sustancia con gran poder fibrinolítico. Esta reacción cuando la desencadenamos farmacológicamente puede ocurrir en todo el territorio circulatorio produciendo un estado de "lisis sistémica", o bien, ocurrir selectivamente a nivel del trombo de fibrina, dependiendo del fármaco empleado. Ninguno de estos fármacos diferencia entre un trombo oclusivo y un coágulo hemostásico, por lo que puede producir hemorragias a cualquier nivel si disuelve uno de estos coágulos.
William R. Flores Ramos 89213
ResponderEliminar1-Activacion plaquetaria
La sangre circula por el interior de los vasos, los glóbulos rojos la más abundante masa celular sanguínea) lo hace por el centro del vaso, mientras que las plaquetas circulan más próximas a la superficie endotelial para vigilar la integridad del endotelio. El endotelio normal proporciona una superficie no adhesiva para las plaquetas. Cuando se rompe el endotelio, el colágeno expuesto favorece la adhesión de las plaquetas por medio de la interacción con una cantidad de receptores,pero es esencial para concretar la adhesión el Factor de Von Willebrand. A partir de esto y con el aumento del Ca++ intracelular se produce la activación de las plaquetas, lo que genera una serie de eventos que favorece la agregación plaquetaria, como son: su cambio de forma y de ser una esfera regular pasa a tener prolongaciones, liberación del contenido de los gránulo como el ADP que torna a las plaquetas pegajosas, el TX A2 derivado del Ácido Araquidónico que es vasoconstrictor y agregante plaquetario. El Factor II (trombina)
2,3,4,5 Coagulacion de la sangre 1,2,3,4
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado. El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc. A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina factor y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos. Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada. La cascada de coagulación se divide en tres vias: La via intrínseca, la vía extrínseca y la vía común.Las vías intrínseca y extrínseca son las vías de iniciación de la cascada, mientras que la vía común es hacia donde confluyen las otras dos desembocando en la conversión de fibrinógeno en fibrina lo cual sella y forma los trombos y coagulos.
6-Trombo
Cuando un vaso sanguíneo se lesiona, el cuerpo utiliza plaquetas y fibrina para formar un coágulo de sangre para prevenir la pérdida de sangre. Incluso cuando un vaso sanguíneo no se lesione, los coágulos de sangre se pueden formar en el cuerpo en ciertas condiciones. El trombo es una masa que se forma en el interior del aparato circulatorio y está constituida por la sangre del paciente, específicamente por los elementos sólidos de la sangre. Las causas son alteración en los vasos sanguíneos como arteriosclerosis,ruptura traumática y alteración en los factores de la coagulación
William R. Flores Ramos 89213
ResponderEliminar7- Fibrinolisis
Es un proceso corporal normal que impide que los coágulos sanguíneos que ocurren en forma natural crezcan y causen problemas.La fibrinólisis primaria se refiere a la descomposición normal de los coágulos.La fibrinólisis secundaria es la descomposición de los coágulos sanguíneos debido a un trastorno médico, un medicamento u otra causa. Esto puede provocar sangrado intenso.
8-Sistema fibrinolitico
La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina(PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón.La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca:En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias,segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA.En la vía intrínseca es la calicreína la encargada de mediar la activación del plasminógeno. La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y elinhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Dubiezel Medina 87865
ResponderEliminarMECANISMOS DE AGREGACION Y ADHERENCIA PLAQUETARIA
Las células endoteliales lesionadas liberan factor de von Willebrand y tromboplastina hística y endotelina (vasoconstrictor potente que reduce la pérdida de sangre).
Las plaquetas se adhieren a la colágena subendotelial, liberan el contenido de sus gránulos y se adhieren unas a otras.
Estos factores en conjunto se conocen como activación plaquetaria.
La liberación de parte de sus contenidos granulares (en especial difosfato de adenosina (ADP) y trombospondina), torna a las plaquetas “pegajosas” y da lugar a que se adhieran las plaquetas circulantes a las plaquetas unidas a la colágena y se desgranulen.
El ácido araquidónico, formado en el plasmalema de plaquetas activadas se convierte en tromboxano A2, un vasoconstrictor y activador de plaquetas potente.
Las plaquetas agregadas actúa como un tapón que bloquea la hemorragia. Además expresan factor 3 plaquetario en su plasmalema, necesario para el ensamble apropiado de factores de la coagulación ( en especial de trombina).
Tanto la tromboplastina hística como la tromboplastina plaquetaria actúa convirtiendo la protombina en trombina, la cual facilita la agregación plaquetaria.
En presencia de calcio también convierte el fibrinógeno en fibrina.
Los monómeros de fibrina que se producen forma un retículo de coágulo que conjunta plaquetas adicionales, eritrocitos y leucocitos en un coágulo sanguíneo (trombo).
Alrededor de una hora después de formarse el coágulo los monómeros de actina y miosina forman filamentos delgados y gruesos , con lo cual se contrae el coágulo casi la mitad de su tamaño previo, tracciona los bordes del vaso, los acerca entre sí y minimiza la pérdida de sangre.
Finalmente cuando se repara el vaso, las células endoteliales liberan activadores del plasminógeno que convierten el plasminógeno circulante en plasmina, la cual inicia la lisis del trombo.
Dubiezel Medina 87865
ResponderEliminarMECANISMOS DE AGREGACION Y ADHERENCIA PLAQUETARIA
Las células endoteliales lesionadas liberan factor de von Willebrand y tromboplastina hística y endotelina (vasoconstrictor potente que reduce la pérdida de sangre).
Las plaquetas se adhieren a la colágena subendotelial, liberan el contenido de sus gránulos y se adhieren unas a otras.
Estos factores en conjunto se conocen como activación plaquetaria.
La liberación de parte de sus contenidos granulares (en especial difosfato de adenosina (ADP) y trombospondina), torna a las plaquetas “pegajosas” y da lugar a que se adhieran las plaquetas circulantes a las plaquetas unidas a la colágena y se desgranulen.
El ácido araquidónico, formado en el plasmalema de plaquetas activadas se convierte en tromboxano A2, un vasoconstrictor y activador de plaquetas potente.
Las plaquetas agregadas actúa como un tapón que bloquea la hemorragia. Además expresan factor 3 plaquetario en su plasmalema, necesario para el ensamble apropiado de factores de la coagulación ( en especial de trombina).
Tanto la tromboplastina hística como la tromboplastina plaquetaria actúa convirtiendo la protombina en trombina, la cual facilita la agregación plaquetaria.
En presencia de calcio también convierte el fibrinógeno en fibrina.
Los monómeros de fibrina que se producen forma un retículo de coágulo que conjunta plaquetas adicionales, eritrocitos y leucocitos en un coágulo sanguíneo (trombo).
Alrededor de una hora después de formarse el coágulo los monómeros de actina y miosina forman filamentos delgados y gruesos , con lo cual se contrae el coágulo casi la mitad de su tamaño previo, tracciona los bordes del vaso, los acerca entre sí y minimiza la pérdida de sangre.
Finalmente cuando se repara el vaso, las células endoteliales liberan activadores del plasminógeno que convierten el plasminógeno circulante en plasmina, la cual inicia la lisis del trombo.
Dubiezel Medina 87865
EliminarFORMACIÓN DEL TROMBO BLANCO
Una vez lesionado el vaso, las plaquetas se adhieren al sitio dañado, que suele ser la superficie de la íntima al descubierto. La adherencia plaquetaria es mediada fundamentalmente por el factor de von Willebrand (von Willebrand factor, vWF), una gran proteína multimérica que está en el plasma y en la matriz extracelular de la pared subendotelial de los vasos, que actúa como el "adhesivo molecular primario", y genera potencia suficiente para soportar las grandes fuerzas de cizallamiento que actuarían para desprenderla con la corriente de sangre. La adherencia mencionada también es facilitada por la unión directa al colágeno subendotelial por los receptores específicos de dicha sustancia en la membrana plaquetaria.
La adherencia plaquetaria origina activación y agregación de los trombocitos; dicho proceso es intensificado y amplificado por mediadores humorales en el plasma (como la adrenalina y la trombina); mediadores liberados de las plaquetas activadas (como el difosfato de adenosina y la serotonina) y los constituyentes de la matriz extracelular de la pared del vaso que se ponen en contacto con las plaquetas adherentes (como colágeno o vWF). Las plaquetas activadas pasan por una reacción de liberación en la cual secretan su contenido, que estimula aún más la agregación e inhibe los factores naturales anticoagulantes de las células del endotelio. En la agregación (interacción entre una plaqueta y otra) hay reclutamiento de más plaquetas desde la circulación hasta el sitio de la lesión vascular, con la cual se forma el llamado trombo blanco oclusivo, hecho de plaquetas, fijado y estabilizado con la trama de fibrina en evolución.
El complejo de glucoproteína plaquetaria (Gp) IIb/IIIa (IIb3) es el receptor más abundante en la superficie de la plaqueta. La activación de los trombocitos transforma al receptor GpIIb/IIIa normalmente inactivo, en otra estructura activa que permite la unión al fibrinógeno y a vWF. La superficie de la plaqueta tiene aproximadamente 50 000 sitios de unión de fibrinógeno GpIIb/IIIa y por ello las innumerables plaquetas activadas reclutadas en el sitio de la lesión vascular rápidamente forman un "agregado" oclusivo por medio de una red densa de puentes de fibrinógeno intercelulares. El receptor en cuestión es el mediador básico de la agregación plaquetaria y por ello se ha vuelto un "elemento" eficaz para ser modificado por medio de terapia antiplaquetaria.
Dubiezel Medina 87865
ResponderEliminarFORMACIÓN DEL COÁGULO DE FIBRINA
Las proteínas de coagulación (factores de coagulación) circulan normalmente inactivas en el plasma. La serie de reacciones de coagulación que culminan en la formación de fibrina fue descrita originalmente como "cascada". Ya se describieron dos vías de coagulación de la sangre hace tiempo: la llamada vía extrínseca o de factor hístico, y la intrínseca o de activación por contacto. Se sabe ahora que la coagulación es iniciada normalmente al exponerse y activarse el factor hístico (tissue factor, TF), siguiendo la vía extrínseca clásica, pero interviene la amplificación importantísima por la participación de elementos de la vía intrínseca clásica. Dichas reacciones acaecen en las superficies fosfolípidas, por lo común en el exterior de la plaqueta activada. Los métodos para evaluar la coagulación en el laboratorio reflejan otros factores de influencia, por la naturaleza artificial de los sistemas in vitro usados.
El elemento desencadenante inmediato de la coagulación es la lesión vascular, que expone la sangre al factor hístico (TF) que es expresado en forma constitutiva en las superficies de los componentes celulares del subendotelio de la pared, como las células de fibra lisa y los fibroblastos. El TF también aparece en micropartículas circulantes quizá desprendidas de células como los monocitos y las plaquetas. Dicho factor se liga al factor VIIa de proteasa serínica; el complejo activa el factor X y se transforma en Xa. Como otra posibilidad, el complejo indirectamente activa el factor X e inicialmente transforma el factor IX en IXa, y como paso siguiente activa el factor X. La participación del factor XI en la hemostasia no depende de su activación por el factor XIIa sino más bien de su activación retroalimentaria positiva por parte de la trombina. De este modo, el factor XIa actúa en la propagación y amplificación y no en el desencadenamiento de la cascada de la coagulación.
El factor Xa, que se forma por las acciones del complejo de factor hístico/factor VIIa o el factor IXa (con el factor VIIIa como cofactor), transforma la protrombina en trombina, que es la proteasa fundamental del sistema de coagulación. El cofactor esencial de dicha reacción es Va. A semejanza de su homólogo el factor VIIIa, el factor Va es producido por la proteólisis limitada del factor V, inducida por trombina. La trombina es una enzima multifuncional que transforma el fibrinógeno plasmático soluble en una matriz insoluble de fibrina. La polimerización de la fibrina entraña un fenómeno ordenado de vínculos intermoleculares. La trombina también activa el factor XIII (factor estabilizante de fibrina) hasta la forma de factor XIIIa, que se enlaza en forma covalente y con ello estabiliza el coágulo de fibrina.
El "ensamblado" de los factores de coagulación en las superficies activadas de la membrana celular acelera enormemente las reacciones que se suceden, y localiza la coagulación en sitios de lesión vascular. Los fosfolípidos ácidos, componentes fundamentales de la membrana celular, no están expuestos normalmente a las superficies de la membrana de la célula en reposo. Sin embargo, cuando la lesión vascular o estímulos inflamatorios activan plaquetas, monocitos y células endoteliales, los grupos "superiores" procoagulantes de los fosfolípidos aniónicos de la membrana son transpuestos a las superficies de tales células o liberados como parte de micropartículas, y quedan disponibles para apoyar y estimular las reacciones de coagulación plasmáticas.
Dubiezel Medina 87865
ResponderEliminarEL SISTEMA FIBRINOLÍTICO
La trombina que "escapa" a los efectos inhibidores de los sistemas anticoagulantes fisiológicos queda disponible para transformar el fibrinógeno en fibrina. En reacción a tal situación el sistema fibrinolítico endógeno es activado para "utilizar" la fibrina intravascular y con ello conservar o restablecer el libre tránsito de la circulación. De la misma forma que la trombina es la proteasa fundamental del sistema de coagulación, la plasmina lo es en el sistema fibrinolítico, al digerir la fibrina y generar sus productos de degradación.
Los activadores de plasminógeno, que son el activador de tipo hístico (tissue type plasminogen activator, tPA) y el de tipo urocinasa (urokinase type plasminogen activator, uPA), separan Arg560-Val561 unida al plasminógeno, para generar la plasmina, enzima activa. Los sitios de unión lisínicos presentes en la plasmina (y el plasminógeno) permiten que se liguen a la fibrina, de tal manera que la fibrinólisis fisiológica es "fibrinoespecífica". El plasminógeno (por medio de sus sitios de unión lisínicos) y tPA poseen afinidad específica por la fibrina, y por lo tanto se ligan selectivamente a los coágulos. El ensamblado de un complejo ternario que consiste en fibrina, plasminógeno y tPA estimula la interacción localizada entre el plasminógeno y tPA, y acelera enormemente la activación de plasminógeno y su transformación en plasmina. Además, la degradación parcial de la fibrina por parte de la plasmina deja al descubierto nuevos sitios de plasminógeno y de unión con tPA en los residuos lisínicos de la terminación carboxilo de los fragmentos de fibrina, para intensificar todavía más tales reacciones. De esa manera, el organismo cuenta con un mecanismo extraordinariamente eficiente para generar en forma focal plasmina en el coágulo de fibrina, y de ese modo se transforma en sustrato de plasmina para la digestión, hasta la aparición de productos de degradación de la fibrina.
La plasmina degrada la fibrina en sitios precisos de su molécula, de modo que se generan los característicos fragmentos de fibrina en el proceso de fibrinólisis. Los sitios de degradación son iguales que los correspondientes al fibrinógeno. Sin embargo, cuando la plasmina actúa en la fibrina con uniones covalentes, quedan en libertad los D-dímeros; en consecuencia, es posible medirlos en el plasma como un "índice" o reflejo relativamente específico de la degradación de fibrina (en vez de medirlos con fibrinógeno). Las mediciones de los D-dímeros se pueden utilizar como marcadores sensibles de la formación de coágulos, y algunos han sido validados para empleo en clínica, para así descartar el diagnóstico de trombosis venosa profunda (deep venous thrombosis, DVT) y embolia pulmonar en poblaciones escogidas.
La regulación fisiológica de la fibrinólisis acaece más bien en dos niveles: 1) inhibidores del activador de plasminógeno (plasminogen activator inhibitors, PAI) en particular PAI1 y PAI2, con inhibición de los activadores de plasminógeno fisiológicos y 2) la antiplasmina 2 inhibe la plasmina. El PAI1 es el inhibidor primario de tPA y uPA en el plasma. La antiplasmina 2 es el principal inhibidor de la plasmina en el plasma de humanos e inactiva cualquier plasmina no vinculada con el coágulo de fibrina.
Dubiezel Medina 87865
ResponderEliminarVIA INTRINSECA
Se inicia por exposición del F. XII a superficies cargadas negativamente (membrana basal pared vaso). El factor XIIa activa la precalicreína a calicreína, que es un potente activador del factor XII (retroalimentación positiva). El F. XIIa, en la misma superficie, activa al factor XI. Estas dos reacciones requieren un cofactor no enzimático: el quininógeno de alto peso molecular (HMWK) que circula unido al factor XI y a la precalicreína. Finalmente el factor XIa activa al factor IX. Todas estas reacciones no requieren iones calcio.
Complejo procoagulante “tenasa”
El factor IXa puede activar al factor X, pero de manera muy lenta. Requiere la presencia de su cofactor, el factor VIIIa. La activación del factor VIII la lleva a cabo la trombina. Una vez formado el VIIIa se genera el complejo tenasa entre el factor VIIIa(cofactor), el IXa (enzima) y el X (sustrato), sobre la superficie fosfolipídica cargada negativamente. Los tres factores se asientan sobre dicha superficie y exponen sus centros activos permitiendo una óptima activación del factor X. Esta superficie la aportan tanto plaquetas activadas como el endotelio dañado ( lo que localiza el coágulo en el sitio de rotura y controla su extensión).
Complejo “protrombinasa”
A continuación el factor Xa (enzima) es liberado y se une al factor Va (cofactor), también activado por la trombina, y al factor II o protrombina (sustrato) generándose el complejo protrombinasa, necesitándose también la presencia de iones calcio y fosfolípidos.
Complejo procoagulante “tenasa extrínseca”
La vía extrínseca se inicia tras el contacto con el factor tisular (FT) que no está normalmente expuesto al torrente circulatorio y que se libera como resultado de un daño endotelial. Ésta es la vía más relevante desde un punto de vista fisiopatológico. El FT se une al factor VII activándolo. El complejo FT-VIIa es capaz de generar trazas de factor Xa. Con lo que el complejo iniciador de la vía extrínseca es el formado por el factor VIIa (enzima), FT (cofactor), factor X (sustrato), iones calcio y una superficie fosfolipídica
Dubiezel Medina 87865
ResponderEliminarActivación de los factores IX y X por el complejo factor VIIa-FT
Sin embargo este proceso es relativamente lento y el complejo factor VIIa-FT-X también es capaz de activar al factor IX (vía intrínseca) con el fin de acelerar la formación del factor Xa. Por tanto el factor Xa es generado por dos vías diferentes. La 1ª, iniciada por el complejo FTVIIa, es más lenta, y ocurre en las primeras etapas, permitiendo la generación de trazas de trombina, que activará al factor VIII, permitiendo la formación del complejo tenasa y acelerando la producción de trombina. Por eso los factores VIII y IX son necesarios para una eficaz coagulación y por lo que las personas con déficits de estos factores sangran (hemofilia. El factor Xa se libera del complejo tenasa y se une al complejo protrombinasa, generando al factor IIa o trombina
VÍA COMÚN: trombina
Además, la trombina activa a los factores V y VIII, a las plaquetas y al factor XIII, que se encarga de estabilizar el coágulo de fibrina. Y, además activa al factor XI, mediante una serie de reacciones de retroalimentación positiva. Al mismo tiempo, la trombina inicia también una de las vías anticoagulantes más potentes, la vía de la proteína C, lo que reduce la propia formación de trombina (retroalimentación negativa).
VÍA COMÚN: fibrinógeno
Molécula formada por 2 subunidades idénticas. Cada subunidad formada por 3 cadenas glicoproteicas: Aα, Bβ, γ. La trombina libera un fragmento de las cadenas Aα y Bβ generando el monómero de fibrina y los fibrinopéptidos A y B. Ello genera cambios estructurales en el fibrinógeno que inducen su polimerización, tanto lineal como lateral, formando una malla tridimensional de fibrina inestable, que estabiliza el factor XIIIa (circula unido al fibrinógeno y es estimulado por la liberación de FPA y FPB). En la red de fibrina quedan también atrapadas células sanguíneas que hacen más impermeable al coágulo.
Dubiezel Medina 87865
ResponderEliminarFibrinolisis
La fibrinolisis es un mecanismo esencial para eliminar los coágulos de fibrina durante el proceso de cicatrización, así como remover los coágulos intravasculares para impedir la trombosis. El efector final del sistema es la plasmina, que degrada la fibrina en productos de degradación (PDF y dímero D). La plasmina es producida a partir de un precursor inactivo, el plasminógeno, por acción de dos activadores del plasminógeno: activador tisular (t-PA) y activador tipo urocinasa (u-PA). La regulación de los activadores tiene lugar por la acción de inhibidores (PAI), de los que el más relevante es el PAI-1, mientras que la plasmina circulante es rápidamente inhibida por la α2-antiplasmina, lo que evita una fibrinolisis sistémica. La fibrinolisis se inicia por el t-PA liberado desde el endotelio en respuesta a diversos estímulos (trombina, oclusión venosa, ejercicio físico, etc). Una vez liberado se une a la fibrina donde activa el plasminógeno a plasmina que degrada la fibrina del coágulo. La trombina puede activar otro inhibidor fibrinolítico, el TAFI, el cual elimina residuos de lisina de la fibrina, lo que impide la unión del plasminógeno y ulterior degradación del coágulo.
dhamelis feliz santana 84917
ResponderEliminarActivación plaquetaria
Un enorme número de sustancias puede actuar sobre las plaquetas e inducir su activación [el factor de activación plaquetaria,6 trombina, el tromboxano (TXA2), fosfato de adenosina (ADP),10 epinefrina, vasopresina y serotonina] a través de sus respectivos receptores, pero todas lo hacen a merced de rutas de señalización intracelular que involucran un sistema de proteínas G acoplado a enzimas efectoras comolas fofolipasas A2 (PLA2) y C (PLC) y adenilato y guanilato ciclasas.5, 6, 10
Hasta ahora se han identificado nueve componentes diferentes de la familia de proteínas G en las plaquetas y, dependiendo del tipo de proteína G que participe en la señalización. Se puede activar o inhibir el efecto.6 Un ejemplo lo constituye la regulación por proteína G de la adenilato ciclasa y por tanto de la producción de monofosfato de adenosina cíclico (AMPc). Un incremento de las cantidades de AMPc bloquea la liberación de calcio en el STD e inhibe la función de la plaqueta. Este es el mecanismo que utilizan las prostaciclinas del endotelio vascular sano para inhibir la activación plaquetaria.
Los procesos de activación plaquetaria comprenden complejas rutas de señalización que involucran sistemas de proteínas G acoplados a enzimas efectoras como la fosfolipasa A2, fosfolipasa C, adenilato y guanilato ciclasas. Las propiedades funcionales y estructurales de las plaquetas están estrechamente relacionadas, lo que les confiere un alto potencial trombogénico, contribuyendo sustancialmente en el desarrollo de un infarto cerebral. Se realizó una revisión de las bases de la agregación, activación, degranulación plaquetaria y su participación en la enfermedad cerebrovascular. Se realizó una actualización del uso de los agentes antiplaquetarios y de la literatura actual referente al uso de bloqueadores de las glicoproteínas adhesivas de membrana
Karina Ortiz del Valle 87546
ResponderEliminarSon muchos los mediadores químicos relacionados con procesos inmunoinflamatorios y que desempeñan un papel importante tanto en afecciones locales como sistémicas.
De manera práctica podemos dividirlos en:
•Sustancias vasoactivas: bradiquinina, histamina, serotonina, etc. (Son mediadores preformados).
•Derivados de fosfolípidos de membrana: prostaglandinas, leucotrienos y factor de activación plaquetaria (FAP). Estos son mediadores neoformados.
•Productos de origen lisosomal: proteasas y proteínas catiónicas implicadas en la destrucción celular.
• Radicales libres: anión superóxido y otros relacionados con la destrucción cellular.
Como toda molécula presente en los seres vivos, los radicales libres ejercen efectos útiles y efectos nocivos. Su papel de defensa durante la fagocitosis es positivo, la amplificación de estos fenómenos en la inflamación, trombosis o isquemia resulta perjudicial para las propias células y tejidos involucrados.
El factor de activación plaquetaria (FAP) es un potente mediador químico de la inflamación, es un derivado fosfolipídico involucrado en múltiples procesos patológicos relacionados con agregación plaquetaria, reacciones inmunoinflamatorias, trastornos vasculares y otros.
La biosíntesis del FAP se produce a partir de los ésteres de glicerilo de las membranas (glicerofosfolípidos). Mediante la fosfolipasa A2 (PLA2) se elimina el residuo acilo y se esterifica el alcohol en 2 (a menudo se trata de un ácido araquidónico), y queda conformado el liso-FAP. Una enzima del grupo de las transacetilasas, que utiliza la acetil coenzima A (acetil CoA), fija el acetilo en 2, y queda conformado el FAP que resulta muy activo y de vida extremadamente corta.
El liso-FAP es a la vez un precursor y un producto de degradación del FAP. No tiene éster acético fijado al alcohol 2 del glicerol y por lo tanto es inactivo. Parece que el liso -FAP puede circular de una célula a otra; por ejemplo, ir de las plaquetas a los neutrófilos y servir de precursor en la biosíntesis de FAP en estas células.
Este comentario ha sido eliminado por el autor.
ResponderEliminarKarina Ortiz 87546
ResponderEliminarCoagulacion 1
El proceso hemostático comprende una fase relacionada con el vaso sanguíneo, otra con las plaquetas y otra en la que se produce la coagulación; se comprende que, para coagular la sangre, se precisa, ante todo, que la corriente sanguínea disminuya su propia velocidad y que la porción de pared de vaso lesionada quede como aislada, que se realice un cierre provisional (fase de las plaquetás) sobre la lesión, y que, por último, se establezca el definitivo (coagulación).
La capa de células que tapiza la superficie interna de los vasos se llama endotelio. En condiciones normales, se distribuyen por él cargas eléctricas negativas y, como también la superficie de las plaquetas está cargada negativamente, son rechazadas por la superficie interna de los vasos, repeliéndose entre sí. Cuando se lesiona un pequeño vaso (si son afectados los grandes vasos la teoría ya no tiene valor, puesto que la sangre sale con fuerza, debido a la elevada presión que existe en ellos, y entonces hay que detener la hemorragia artificialmente), como en el caso de una herida trivial, se produce una modificación de la carga eléctrica del endotelio en el punto lesionado. Entonces, las plaquetas se acumulan en gran número en este punto y se adhieren a la porción de superficie lesionada (donde se ha modificado el potencial eléctrico), formando un tapón, que cierra el punto abierto por el que escapa la sangre. Simultáneamente, la presión en el organismo desciende y disminuye también la fuerza con que la sangre escapa al exterior. Una vez que las plaquetas han entrado en contacto con la zona lesionada, empiezan a adelgazarse es decir, a disminuir de espesor, y a extenderse aumentando su diámetro. Paralelamente a este fenómeno, las plaquetas comienzan a desintegrarse. Mediante su destrucción se liberan diversas sustancias, entre las cuales está la serotonina, que provoca rápidamente una contracción de los vasos lesionados y de los que los rodean próximamente: de este modo, la sangría se estanca. La serotonina es una sustancia producida por unas células especiales del intestino, denoinadas células enterocromafinas; pasa luego a la corriente sanguínea, donde es absorbida por las plaquetas y liberada por éstas cuando se desintegran. Hay también otra sustancia (factor plaquetario) que, en presencia del calcio contenido habitualmente en el plasma contribuye a iniciar el proceso de coagulación.
Pero el coágulo, después de haberse formado, es todavía blando y friable, hasta que se produce una retracción, que lo transforma en una masa más compacta.
Karina Ortiz 87546
ResponderEliminarCoagulacion 2
La coagulación es, en cierto sentido, un mecanismo de defensa de enorme utilidad para el organismo. En condiciones normales, tiene la misión concreta de limitar, hasta detenerlas, las pérdidas de sangre debidas a eventuales lesiones de los vasos sanguíneos. Este dispositivo, muy delicado y complejo, está confiado a unos factores que se encuentran en el plasma, y a las plaquetas, unos componentes celulares que figuran entre los "elementos corpusculares" de la sangre. En realidad, las plaquetas no son células, sino, simplemente, fragmentos de citoplasma (sin núcleo) de dos o tres milésimas de milímetro de diámetro, que se desprenden de células muy grandes, llamadas megacariocitos. Los megacariocitos residen en la médula ósea y, habitualmente, no penetran en la circulación; sólo los trocitos de citoplasma que se desprenden de ellos son transportados por la sangre. Las plaquetas sobreviven tres días o poco más, por término medio, y luego, si entre tanto no han sido utilizadas en el proceso de detención de una eventual hemorragia, sufren el mismo destino de los demás elementos de la sangre, es decir, son destruidos por las células del "sistema reticuloendotelial".
Los factores del plasma que entran en el mecanismo de la coagulación están representados por el fibrinógeno, la protrombina, el calcio y otras numerosas sustancias. Pero la coagulación no sería suficiente por sí sola para detener la salida de sangre de la lesión de un vaso. Aunque, en realidad, es el mecanismo más importante y perfeccionado, forma parte de un conjunto de fenómenos, orientados todos a detener la hemorragia, que toma el nombre de hemostasia.
Karina Ortiz 87546
ResponderEliminarCoagulación 3
El proceso de coagulación más propiamente dicho, consiste en la transformación de una sustancia presente en el plasma, llamada fibrinógeno, en otra llamada fibrina. Esta reacción se produce por obra de la trombina, que se forma a partir de la protrombina, bajo la influencia de la tromboplastina. En condiciones normales, no existe trombina (pero está presente un precursor inactivo suyo, la protrombina, que es una proteína de origen hepático) ni tromboplastina, pues de lo contrario, la sangre se coagularía habitualmente en los vasos con graves consecuencias.
Por lo tanto, en la coagulación podemos distinguir tres momentos:
•1) formación de la tromboplastina
• 2) transformación de la protrombina en trombina
•3) transformación del fibrinógeno, proteína soluble en fibrina insoluble.
La formación de la tromboplastina es muy compleja; se debe a la activación de diversos factores presentes en el plasma y en los tejidos; la activación de uno de ellos repercute sobre el inmediatamente siguiente no activado todavía, como en una reacción en cadena.
El punto de partida de este proceso es doble: es decir, el proceso es puesto en marcha tanto por factores contenidos en el plasma, como por otros presentes en los tejidos (factores plasmáticos y factores tisulares), que se liberan a raíz de la salida de la sangre. Una vez formada, la tromboplastina actúa rápidamente sobre la protrombina y la transforma en trombina; de este modo, termina también la segunda fase de la coagulación y comienza la tercera, en la que la trombina actúa sigue y lo transforma en fibrina, sustancia, por el contrario, insoluble en el plasma (parte líquida de la sangre). El fibrinógeno es una gruesa proteína en forma de agujas, que se modifica bajo la acción de la trombina. Muchos elementos de fibrinógeno así modificados se unen por los extremos, dando lugar a larguísimos filamentos muy delgados. Éstos se reúnen en haces de mayor espesor, que se agrupan entre sí, formando una densa red visible de fibrina, entre cuyas mallas se encuentran aprisionados glóbulos rojos, glóbulos blancos y restos de plaquetas. Este es el coágulo.
Karina Ortiz 87546
ResponderEliminarCoagulacion 4
Existen dos vías llevan a la formación de un coágulo de fibrina: la vía intrínseca y la vía extrínseca. Aunque las dos son iniciadas por mecanismos diferentes, las dos convergen en una vía común que lleva a la formación del coágulo. La formación del trombo rojo ó coágulo en respuesta a una anormalidad en un vaso pero en la ausencia de una lesión del tejido es el resultado de la vía intrínseca.
La vía intrínseca tiene poca significancia bajo condiciones fisiológicas normales. El suceso más importante clínicamente es la activación de la vía intrínseca por el contacto de la pared del vaso con las partículas de lipoproteína, VLDLs y quilomicrones. Este proceso claramente demuestra el papel de la hiperlipidemia en la generación de la ateroesclerosis. La vía intrínseca también puede ser activada por el contacto de la pared del vaso con bacterias.
La formación del coágulo de fibrina en respuesta a una lesión del tejido es el evento clínico más importante de la hemostasis bajo condiciones normales de respuesta. Este proceso es el resultado de la activación de la vía extrínseca.
Ambas vías son complejas e incluyen varias proteínas diferentes denominadas factores de la coagulación.
Karina Ortiz 87546
ResponderEliminarTrombo y Coágulo
Un trombo o coágulo de la sangre, es el producto final de la etapa de coagulación de la sangre en la hemostasis. Esto se logra a través de la agregación de las plaquetas que forman un tapón de plaquetas, y la activación del sistema de coagulación humoral (es decir, los factores de coagulación). Un trombo es normal en los casos de lesiones, pero patológica en casos de trombosis.
Son los trombos murales trombos adherente a la pared del vaso. Ellos no son oclusivos y afectan a los vasos grandes, tales como el corazón y la aorta. Macroscópicamente que aparecen en gris-rojo con líneas claras y oscuras (líneas de Zahn) que representan bandas de fibrina (más ligero) con las células blancas de la sangre atrapados y las células rojas de la sangre (más oscuro) alterna.
En concreto, un trombo es la activación inapropiada del proceso hemostático en un recipiente sin lesiones o heridas leves. Un trombo en un vaso sanguíneo grande disminuirán el flujo de sangre a través de ese recipiente (denominado un trombo mural). En un pequeño vaso sanguíneo, el flujo sanguíneo puede ser completamente de corte (denominado un trombo oclusivo) que resulta en la muerte de tejido suministrado por dicho buque. Si se desplaza un trombo y se convierte en libre flotación, que se denomina como un émbolo.
Algunas de las condiciones que elevan el riesgo de coágulos sanguíneos en desarrollo incluyen la fibrilación auricular (un tipo de arritmia cardiaca), reemplazo de válvula cardíaca, un reciente ataque al corazón (también llamado infarto de miocardio), períodos largos de inactividad, y deficiencias genéticas o enfermedades relacionadas en las capacidades de coagulación de la sangre.
87462 ESTEBAN M GUERRERO P
ResponderEliminarDIAPOSITIVAS TODAS
La habilidad del cuerpo para controlar el flujo de sangre luego de una lesión vascular es un componente indispensable de la supervivencia. El proceso de la coagulación sanguínea y luego la disolución del coágulo, seguido por una reparación del tejido lesionado, se denomina hemostasis. La hemostasis se conforma de 4 eventos principales que ocurren en un orden determinado luego de la pérdida de la integridad vascular:
1. La fase inicial del proceso es la constricción vascular. Esto limita el flujo sanguíneo al área de la lesión.
2. A continuación, se activan las plaquetas por la trombina y se agregan en el sitio de la lesión, formando un tampón temporario y flojo conformado de plaquetas. La proteína fibrinógeno es principalmente responsable de estimular la agregación plaquetaria. Las plaquetas se agregan al unirse al colágeno que se expone debido a la ruptura del recubrimiento epitelial de los vasos. Luego de su activación, las plaquetas liberan ADP un nucleótido y un eicosanoide, TXA2 (los cuales activan más plaquetas), serotonina, fosfolípidos, lipoproteínas y otras proteínas importantes de la cascada de coagulación. Además de su secreción, las plaquetas activadas cambian su conformación para acomodar la formación del coágulo.
3. Para asegurar la estabilidad del tampón flojo inicial, se forma una malla de fibrina (también llamada un coágulo) que recubre al tampón. Si el tampón únicamente contiene plaquetas se denomina un trombo blanco; si glóbulos rojos están presentes se lo denomina un trombo rojo
4. Finalmente, el coágulo debe ser disuelto para que el flujo sanguíneo normal pueda resumir luego de que se repare el tejido. La disolución del coágulo ocurre a través de la acción de la plasmina
Dos vías llevan a la formación de un coágulo de fibrina: la vía intrínseca y la vía extrínseca. Aunque las dos son iniciadas por mecanismos diferentes, las dos convergen en una vía común que lleva a la formación del coágulo. La formación del trombo rojo ó coágulo en respuesta a una anormalidad en un vaso pero en la ausencia de una lesión del tejido es el resultado de la vía intrínseca. La vía intrínseca tiene poca significancia bajo condiciones fisiológicas normales. El suceso más importante clínicamente es la activación de la vía intrínseca por el contacto de la pared del vaso con las partículas de lipoproteína, VLDLs y quilomicrones. Este proceso claramente demuestra el papel de la hiperlipidemia en la generación de la ateroesclerosis. La vía intrínseca también puede ser activada por el contacto de la pared del vaso con bacterias.
La formación del coágulo de fibrina en respuesta a una lesión del tejido es el evento clínico más importante de la hemostasis bajo condiciones normales de respuesta. Este proceso es el resultado de la activación de la vía extrínseca.
Ambas vías son complejas e incluyen varias proteínas diferentes denominadas factores de la coagulación.
87462 ESTEBAN M GUERRERO P
ResponderEliminarDIAPOSITIVAS TODAS
Activación Plaquetaria y el Factor von Willebrand (vWF)
Para que ocurra la hemostasis, las plaquetas deben adherirse al colágeno expuesto, liberar los contenidos de sus gránulos y agregarse. La adhesión plaquetaria al colágeno expuesto en las superficies endoteliales de las células es mediada por el factor von Willebrand (vWF). Las deficiencias hereditarias del vWF son la causa de la enfermedad de von Willebrand, (vWD) (ver abajo para más detalles). La función del vWF is actuar como un puente entre un complejo específico de glicoproteínas en la superficie de las plaquetas (GPIb-GPIX-GPV) y las fibrillas de colágeno. El GPIb parte del complejo se compone de dos proteínas, GPIbα y GPIbβ codificadas por los genes por separado. La importancia de esta interacción entre el vWF y el complejo GPIb de las plaquetas es ilustrado en los desórdenes hereditarios de la coagulación causados por defectos en las proteínas del complejo GPIb, de los cuales el más común es el síndrome de Bernard-Soulier (también conocido como el síndrome de las plaquetas gigantes).
Además de servir como un puente entre las plaquetas y el colágeno expuesto de las superficies endoteliales, el vWF se une y estabiliza el factor de coagulación VIII. La unión del factor VIII por el vWF es necesaria para la supervivencia normal del factor VIII en la circulación.
El factor von Willebrand es una glicoproteína multimérica compleja que es producida y almacenada en los gránulos α de las plaquetas. También es sintetizado por los megacariocitos y se encuentra asociado al tejido conectivo subendotelial.
La activación inicial de las plaquetas es inducida por una trombina ligada a un receptor específico ubicado en la superficie de las plaquetas, lo cual inicia la transducción de una cascada de señalización. El receptor de la trombina está acoplado a una proteína G que a su vez activa a la fosfolipasa C-γ (PLC-γ). La PLC-γ hidroliza al fosfatidilinositol-4,5-bifosfato (PIP2) resultando en la formación de inositol trifosfato (IP3) y diacilglicerol (DAG). El IP3 induce la liberación de los almacenes intracelulares de Ca2+ y el DAG activa a la proteína cinasa C (PKC).
El colágeno al cual se adhieren las plaquetas al igual que la liberación del Ca2+ intracelular resulta en la activación de la fosfolipasa A2 (PLA2) la cual a continuación hidroliza fosfolípidos en la membrana, llevando a la liberación del ácido araquidónico. La liberación del ácido araquidónico resulta en un aumento en la producción y liberación del tromboxano A2 (TXA2). El TXA2 es un vasoconstrictor potente e induce la agregación plaquetaria y funciona al unirse a receptores que trabajan a través de la vía de la PLC-γ.
Otra enzima que es activada por la liberación del Ca2+ intracelular es la cinasa de la cadena liviana de miosina myosin light chain kinase (MLCK). La MLCK activada fosforila a la cadena liviana de la miosina la cual interactúa con la actina, resultando en una alteración en la morfología y motilidad plaquetaria.
Uno de los varios efectos de la PKC es la fosforilación y activación de una proteína plaquetaria específica de 47,000 Daltons. Esta proteína activada induce la liberación de los contenidos de los gránulos plaquetarios; uno de los cuales es el ADP. El ADP promueve la estimulación plaquetaria al incrementar la activación general de la cascada. El papel importante del ADP en la activación plaquetaria puede apreciarse con el uso de antagonistas del receptor del ADP como el Plavix® (clopidogrel), en el control de la trombosis (ver abajo).
87462 ESTEBAN M GUERRERO P
ResponderEliminarDIAPOSITIVAS TODAS
El ADP también modifica la membrana de las plaquetas conllevando a una exposición del complejo de receptores de glicoproteínas plaquetarias: GPIIb-GPIIIa. El GPIIb-GPIIIa constituye un receptor para el vWF y el fibrinógeno, resultando en una agregación plaquetaria inducida por el fibrinógeno. El complejo GPIIb-GPIIIa es un miembro de una familia de integrinas de receptores de la superficie celular que interactúan con la matríz extracelular. El complejo GPIIb-GPIIIa también es llamado αIIb-β3 integrina. La importancia del GPIIb-GPIIIa en la activación plaquetaria y la coagulación es ilustrada por el hecho de que los desórdenes de coagulación resultan de defectos heredados en este complejo de glicoproteínas. El más común de estas disfunciones plaquetarias hereditarias es la trombastenia de Glanzmann la cual resulta de defectos en la proteína GPIIb de este complejo. Además, la importancia de este complejo en la hemostasis es demostrada por el uso de anticuerpos que bloquean este receptor y actúan como anti-coagulantes (p. ej. ReoPro®, abciximab: ver abajo).
La activación de las plaquetas es necesaria para su consecuente agregación y formación del coágulo plaquetario. Sin embargo, igualmente de importante en la activación de la cascada de a coagulación es el papel de los fosfolípidos activados en la superficie de las plaquetas.
Las cascadas de coagulación: la cascada intrínseca (la cual tiene menos importancia in vivo en comparación a la cascada extrínseca) es iniciada cuando se realiza el contacto entre la sangre y las superficies expuestas de carga negativa. La vía extrínseca es iniciada cuando ocurre una lesión vascular lo cual resulta en una exposición del factor tisular (TF) (también identificado como el factor III), una glicoproteína subendotelial en la superficie celular que se une a fosfolípidos. La flecha punteada verde representa un punto de cruce entre la vía extrínseca y la intrínseca. Las dos vías se convergen en la activación del factor X a Xa. El factor Xa tiene un papel en la activación del factor VII a VIIa como se demuestra por la flecha verde. El factor Xa activo hidroliza y activa la protrombina a trombina. La trombina puede por ende activar los factores XI, VIII y V lo cual ayuda a que proceda la cascada. Básicamente, el papel de la trombina es convertir el fibrinógeno a fibrina y activar el factor XIII a XIIIa. El factor XIIIa (también llamado transglutaminasa) se une a polímeros de fibrina y así solidificando el coágulo. HMWK = quininógeno de alto peso molecular. PK = precalicreina. PL = fosfolípidos.
El Kallikrein-Kinin Sistema de Coagulación
El kallikrein-kinin sistema comprende un complejo de proteínas que, cuando activado conduce a la liberación de kinins vasoactivos. El kinins son liberados de alto peso molecular kininogen (HMWK) y de bajo peso molecular kininogen (LMWK) como consecuencia de la activación de cualquiera de los tejidos kallikrein o plasma kallikrein. El kallekreins sí existen en la pre-formas inactivas. El kinins están involucrados en muchos fisiológicos y patológicos incluidos los procesos de regulación de la presión arterial y el flujo (a través de la modulación de la renina-angiotensina vía), la coagulación de la sangre, la proliferación celular y el crecimiento, la angiogénesis, apoptosis, y la inflamación. Kinin acción en las células endoteliales conduce a la vasodilatación, el aumento de la permeabilidad vascular, la liberación de activador tisular del plasminógeno (tPA), la producción de óxido nítrico (NO), y la movilización de ácido araquidónico, fundamentalmente el resultado de la prostaciclina (PGI2) la producción por las células endoteliales. Aunque las actividades de la kallikrein-kinin sistema están involucrados en numerosos procesos, en esta sección se ocupará sólo con su función en la coagulación de la sangre. Con respecto a la hemostasia más importante es la bradiquinina kinin que se libera de HMWK.
Karina Ortiz 87546
ResponderEliminarLa fibrinólisis es un proceso que previene los coágulos de sangre de crecimiento y convertirse en problemática [1] Este proceso tiene dos tipos:. Fibrinolisis primaria y fibrinólisis secundaria. El tipo principal es un proceso normal del cuerpo, mientras que la fibrinólisis secundaria es la descomposición de los coágulos debido a un medicamento, un trastorno médico, o alguna otra causa.
En la fibrinólisis, un coágulo de fibrina, el producto de la coagulación, se descompone. [2] Su plasmina principal enzima corta la malla de fibrina en varios lugares, que conduce a la producción de fragmentos circulantes que se eliminan por otras proteasas o por el riñón y el hígado .
La plasmina se produce en una forma inactiva, el plasminógeno, en el hígado. Aunque plasminógeno no puede escindir la fibrina, que todavía tiene una afinidad por ella, y se incorpora en el coágulo cuando se formó.
Activador del plasminógeno tisular (t-PA) [3] y uroquinasa son los agentes que convierten el plasminógeno en la plasmina activa, permitiendo de este modo que se produzca la fibrinólisis. t-PA se libera en la sangre muy lentamente por el endotelio dañado de los vasos sanguíneos, de tal manera que, después de varios días (cuando el sangrado se ha detenido), el coágulo se descompone. Esto ocurre porque plasminógeno se convirtió en atrapado dentro del coágulo cuando se formó, ya que se activa lentamente, se descompone la malla de fibrina. t-PA y uroquinasa se sí mismos inhibidas por el inhibidor del activador del plasminógeno-1 y el inhibidor del activador del plasminógeno-2 (PAI-1 y PAI-2). En contraste, la plasmina estimula aún más la generación de plasmina mediante la producción de formas más activas de tanto activador del plasminógeno tisular (tPA) y la uroquinasa.
Alfa 2-antiplasmina y alfa 2-macroglobulina inactivan plasmina. Actividad de la plasmina también se reduce por el inhibidor de la fibrinólisis activable por trombina (TAFI), que modifica la fibrina para que sea más resistente a la del plasminógeno mediada por tPA.
87462 ESTEBAN M GUERRERO P
ResponderEliminarDIAPOSITIVAS TODAS
Las dos formas de prekallikrein, plasma y tejidos, se derivan de diferentes los genes en diferentes cromosomas. El plasma kallikrein gen (símbolo KLKB1) está en el cromosoma 4q34–q35 y el tejido kallikrein gen (símbolo KLK1) situado en el cromosoma 19q13.2–q13.4. Estos dos son serina proteasas kallikreins cuyos principales sustratos son HMWK (plasma kallikrein) y LMWK (tejido kallikrein). Cuándo plasma prekallikrein está activada para kallikrein que Cleaves HMWK en dos fases proceso que, en definitiva, libera bradiquinina. Bradiquinina es un 9-aminoácido péptido vasoactivo que induce a la vasodilatación y el aumento vasculares permeabilidad. Activado tejido kallikrein Cleaves lysyl-bradiquinina (también llamado kallidin) de LMWK. Lysyl-bradiquinina es bradiquinina con un residuo de la lisina en la N-terminal que un 10-aminoácido péptido vasoactivo. Sus actividades son esencialmente idénticos a los de la bradiquinina.
Ambos HMWK y LMWK se derivan del mismo gen en el cromosoma 3q26–qter que está compuesto de 11 exones. Exones 1 al 9 de codificar la cadena pesada de ambas kininogens. Exón 10 codifica bradiquinina, así como la luz de la cadena de HMWK. Exón 11 codifica la cadena de luz LMWK. La pesada cadena de la luz y la nomenclatura se refiere a la servidumbre-disulfuro de la estructura de cada kininogen después de su a través de la activación por kallikrein escisión.
HMWK se considera una α-globulina y se compone de seis áreas funcionales. La proteína que circula en el plasma como una sola cadena polipeptídica con un peso molecular de 88–120 kDa depende el nivel de glucosilación. El cadena pesada es de 64 kDa y contiene los dominios 1, 2 y 3 mientras que la cadena de luz es de 45–56 kDa y comprende dominios 5 y 6. La cadenas pesadas y ligeras son unidos entre sí a través de dominio 4, que también contiene la secuencia de la bradiquinina. Dominio 1 contiene una baja afinidad de unión de calcio. Dominios 2 y 3 contienen secuencias de aminoácidos (QVVAG) que inhiben la cisteína proteasas. Dominio 3 también ha de plaquetas y células endoteliales vinculante actividad. Dominio de las secuencias de 5 ha heparina vinculante, sitios de unión celular, propiedades y antiangiogénicos. La unión de HMWK a las superficies de carga negativa se produce a través de una región de la histidina cadena de luz que se encuentra en dominio 5. 6 contiene el dominio prekallikrein y factor XI-sitios de unión. Al ser capaces de obligar a cargado a través de superficies de dominio 5 y al mismo tiempo obligar factor XI y prekallikrein dominio a través de 6, puede servir como HMWK cofactor en la activación de contacto con el plasma. LMWK se considera una β-globulina y tiene un peso molecular de 50 a 68 kDa. La estructura de LMWK es similar a la que de HMWK, sin embargo, la cadena de luz está a sólo 4–5 kDa y no tiene ningún contacto activación ni prekallikrein-sitios de unión.
El plasma kinin que forman el sistema se llama el sistema de contacto y de plasma se compone de factor XII, factor XI, prekallikrein y HMWK. Factor XII, prekallikrein, y HMWK saturably reversible y se unen a las células endoteliales, plaquetas y granulocitos en una reacción dependiente de zinc. Cuando se hace de plasma contacto con una superficie de carga negativa, el factor XII se une y se autoactivated del factor XII (el "uno" significa el factor activado). Entonces el factor XIIa prekallikrein activa a kallikrein y kallikrein Cleaves HMWK liberación bradiquinina. Existe también la activación del factor de reciprocidad XII por kallikrein resultante en el sistema de amplificación.
87462 ESTEBAN M GUERRERO P
ResponderEliminarDIAPOSITIVAS TODAS
Cascada Intrínseca de la Coagulación
La vía intrínseca (también llamada la vía de activación por contacto) es mucho menos significante en la hemostasis bajo condiciones fisiológicas normales en comparación a la vía extrínseca. Sin embargo, durante un estado patológico tal como la hiperlipidemia o una infiltración bacteriana puede conllevar a la activación de la trombosis a través de la cascada de coagulación de la vía intrínseca.
La vía intrínseca requiere de los factores de coagulación VIII, IX, X, XI y XII. También requiere de proteínas tales como la precalicreina (PK) y el quininógeno de alto peso molecular (HK o HMWK), al igual que iones de calcio y fosfolípidos secretados por las plaquetas. Cada uno de los componentes de estas vías resulta en la conversión del factor X (inactivo) al factor Xa ("a" significa activo). La iniciación de la vía intrínseca ocurre cuando la precalicreina, quininógeno de alto peso molecular, factor XI y el factor XII son expuestos a una superficie de carga negativa. A esto se lo denomina como la fase de contacto y puede ocurrir como el resultado de una interacción con los fosfolípidos (principalmente fosfotidiletanolamina, PE) de las partículas lipoproteínas circulantes tales como los quilomicrones, VLDL y LDL oxidada. Esta es la base del papel de la hiperlipidemia en promover un estado pro-trombótico y en el desarrollo de la arterosclerosis.
La activación de contacto de la vía intrínseca también puede ocurrir en la superficie de las bacterias, y mediante la interacción con el ácido úrico cristales, ácidos grasos, protoporfirina, β amiloide, y homocisteína. De hecho, los niveles elevados de homocisteína en la sangre han demostrado que son equivalentes con disfunción cardiovascular. Por lo tanto, es importante garantizar que la función apropiada de la reacción metionina sintasa se mantiene. Aunque sería suponer que una mayor ingesta de vitamina B12 debería conducir a Conversión de aumento de la homocisteína en metionina, y por lo tanto reduce los niveles de circulantes de homocisteína, estudios controlados han demostrado que esto no ocurrir.
El ensamblaje de los componentes de la fase de contacto resulta en la conversión de la precalicreina a la calicreina, la cual a su vez activa al factor XII a factor XIIa. El factor XIIa puede entonces hidrolizar más precalicreina a calicreina, estableciendo una recíproca activación de la cascada. El factor XIIa también activa al factor XI a factor XIa y conlleva a la liberación de bradicinina, un vasodilatador potente, a partir del quininógeno de alto peso molecular.
En la presencia del Ca2+, el factor XIa activa al factor IX a factor IXa. El factor IX es una proenzima que contiene residuos γ-carboxiglutamato (gla) vitamina K-dependientes, cuya actividad de proteasas de serina es activada luego de que el Ca2+ se une a los residuos gla. Varias de estas proteasas de serina de la cascada (II, VII, IX y X) son proenzimas que contienen gla. El factor activo IXa cliva al factor X en una unión interna de arg-ile (R-I) resultando en su propia activación al factor Xa.
La activación del factor Xa requiere el ensamblaje del complejo de tenasa (Ca2+ y los factores VIIIa, IXa y X) en la superficie de las plaquetas activadas. Una de las respuestas de las plaquetas a su activación es la presentación de un fosfatidilserina (PA) y un fosfatidilinositol (PI) en sus superficies. La exposición de estos fosfolípidos permite que se forme el complejo de tenasa. El papel del factor VIII en este proceso es de servir como un receptor, en la forma del factor VIIIa, para los factores IXa y X. El factor VIIIa se denomina un cofactor en la cascada de coagulación. La activación del factor VIII al factor VIIIa (el verdadero receptor) ocurre en la presencia de cantidades diminutas de trombina.
Karina Ortiz 87546
ResponderEliminarEl sistema fibrinolítico sanguíneo comprende una proenzima inactiva, plasminógeno, que se puede convertir a la enzima activa, plasmina. La plasmina degrada la fibrina en productos de degradación de fibrina solubles, por los dos activadores del plasminógeno fisiológicas (PA), el tipo de tejido PA (t-PA) y el PA de tipo urocinasa (u-PA). mediada por la activación del plasminógeno de t-PA está implicado principalmente en la disolución de la fibrina en la circulación. u-PA se une a un receptor celular específico (u-PAR), resulta en una mayor activación de células del plasminógeno unido. La inhibición del sistema fibrinolítico puede ocurrir ya sea en el nivel de la PA, por los inhibidores de activador de plasminógeno específico (PAI), o en el nivel de la plasmina, principalmente por alfa-2 antiplasmina. Varias interacciones moleculares se han observado entre el sistema de las metaloproteinasas de matriz (MMP) y fibrinolítico, ambos sistemas pueden colaborar en la generación de actividad proteolítica. Por lo tanto, estromelisina-1 (MMP-3) escinde un fragmento de 55 kDa kringle 1-4, que contiene el sitio de unión de lisina (s) implicada en la unión celular, a partir de plasminógeno y elimina un 17-kDa fragmento NH2-terminal, que contiene el celular El sitio de unión al receptor, de uroquinasa (u-PA). De este modo, MMP-3 puede regular a la baja actividad de la plasmina asociada celular por la disminución de la cantidad de plasminógeno activable, sin afectar célula vinculada actividad de u-PA.
87462 ESTEBAN M GUERRERO P
ResponderEliminarDIAPOSITIVAS TODAS
Cascada Extrínseca de la Coagulación
El factor Xa activado es el sitio en el cual las cascadas de coagulación intrínseca y extrínseca se convergen. La vía extrínseca es iniciada en el sitio de la lesión en respuesta a la liberación del factor tisular (factor III) y por ende, es también conocida como la vía del factor tisular. El factor tisular es un cofactor en la activación catalizada del factor X por el factor VIIa. El factor VIIa una proteasa de serina que contiene un residuo gla, cliva al factor X en factor Xa de manera idéntica a la del factor IXa en la vía intrínseca. La activación del factor VII ocurre a través de la acción de la trombina o el factor Xa. La habilidad del factor Xa de activar al factor VII crea una asociación entre las vías intrínseca y extrínseca. Una asociación adicional entre las dos vías se da a través de la habilidad del factor tisular y el factor VIIa de activar al factor IX. Se cree que la formación del complejo entre el factor VIIa y el factor tisular es el paso principal en toda la cascada de coagulación. La evidencia de esta declaración nace del hecho que personas con deficiencias hereditarias en los componentes de la fase de contacto de la vía intrínseca no exhiben problemas de coagulación. Uno de los mecanismos más importantes de la inhibición de la vía extrínseca ocurre en el complejo tisular factor-factor VIIa–Ca2+–Xa. La proteína, un inhibidor de la coagulación asociado a una lipoproteína, LACI se une específicamente a este complejo. LACI también se denomina el inhibidor de la vía extrínseca, EPI o factor tisular inhibidor de la vía, TFPI y anteriormente se lo conocía como anticonvertina. LACI es compuesto de 3 dominios inhibidores de proteasas. El dominio 1 se une al factor Xa y el dominio 2 se une al factor VIIa pero sólo en la presencia del factor Xa.
Regreso al inicio
Activación de la Protrombina a Trombina
El punto en el cual las dos vías se convergen es en la activación del factor X al factor Xa. El factor Xa activa a la protrombina (factor II) para convertirse en trombina (factor IIa). La trombina, a su vez, convierte el fibrinógeno a fibrina. La activación de la trombina ocurre en la superficie de las plaquetas activadas y requiere de la formación de un complejo de protrombinasa. Este complejo está compuesto de los fosfolípidos de las plaquetas: fosfatidilinositol y fosfatidilserina, Ca2+, factores Va y Xa y protrombina. El factor V es un cofactor en la formación del complejo de protrombinasa y tiene un papel similar al del factor VIII en la formación del complejo de tenasa. Como la activación del factor VIII, el factor V es activado al factor Va a través de cantidades diminutas y es inactivado por niveles aumentados de trombina. El factor Va se une a receptores específicos en la superficie de las plaquetas activadas y forma un complejo con la protrombina y el factor Xa.
La protrombina es una proteína de cadena simple de 72,000 Daltons, que contiene diez residuos gla en la región de su N-terminal. Dentro del complejo de protrombinasa, la protrombina es clivada en 2 sitios por el factor Xa. Este clivaje genera una molécula de trombina activa de 2 cadenas que contiene una cadena A y una cadena B las cuales están unidas por una unión simple de disulfuro.
Además de su papel en la activación de la formación del tampón de fibrina, la trombina juega un papel regulador importante en la coagulación. La trombina se une con la trombomodulina presente en la superficie de las células endoteliales formando un complejo que convierte la proteína C a la proteína Ca. La proteína S y la proteína Ca son cofactores que degradan a los factores Va y VIIIa y así limitando la actividad de estos 2 factores en la cascada de la coagulación.
87462 ESTEBAN M GUERRERO P
ResponderEliminarDIAPOSITIVAS TODAS
Activación del Fibrinógeno a Fibrina
El fibrinógeno (factor I) consiste en 3 pares de polipéptidos ([Aα][Bβ][γ])2. Las 6 cadenas están covalentemente unidas cerca de sus N-terminales a través de uniones disulfuro. Las porciones A y B de las cadenas Aα y Bβ constituyen los fibrinopéptidos, A y B, respectivamente. Las regiones de fibrinopéptidos del fibrinógeno contienen varios residuos de glutamato y aspartato impartiendo una alta carga negativa a esta región y promoviendo la solubilidad del fibrinógeno en el plasma. La trombina activa es una proteasa de serina que hidroliza al fibrinógeno en cuatro uniones arg-gly (R-G) entre el fibrinopéptido y las porciones a y b de la proteína.
La liberación de los fibrinopéptidos mediada por la trombina, genera monómeros de fibrina con una estructura de subunidades (αβγ)2. Estos monómeros se agregan espontáneamente en un arreglo regular, formando un coágulo de fibrina relativamente débil. Además de la activación de la fibrina, la trombina convierte el factor XIII al factor XIIIa, una transglutaminasa altamente específica que introduce uniones covalentes entre el nitrógeno de amidas de las glutaminas y el grupo ε-amino de las lisinas pertenecientes a los monómeros de fibrina.
Regreso al inicio
Disolución de los Coágulos de Fibrina
La degradación de los coágulos de fibrina es la función de la plasmina, una proteasa de serina que circula como la proenzima inactiva plasminógeno. Cualquier plasmina libre circulante es rápidamente inhibida por la antiplasmina α2. El plasminógeno se une al fibrinógeno y a la fibrina, y así se incorporan en un coágulo. El activador tisular del plasminógeno (tPA) y, a un grado menor, la urocinasa son proteasas de serina que convierten el plasminógeno a plasmina. El tPA inactivo es liberado de las células endoteliales vasculares luego de una lesión; se une a la fibrina y es consecuentemente activado. La urocinasa es producida como el precursor de la prourocinasa por las células epiteliales que recubren los conductos excretorios. El papel de la urocinasa es activar la disolución de los coágulos de fibrina que pueden depositarse en estos conductos.
La tPA activa cliva al plasminógeno para producir plasmina la cual digiere a la fibrina; esta degradación resulta en un producto soluble al cual ni la plasmina ni el plasminógeno se pueden unir. Luego de la liberación del plasminógeno y la plasmina, éstos se inactivan rápidamente por sus respectivos inhibidores. La inhibición de la actividad de la tPA resulta de la unión de específicas proteínas inhibidoras. Por lo menos 4 diferentes inhibidores han sido identificados, dos de los cuales son inhibidores de los activadores del plasminógeno tipo I (PAI-1) y tipo 2 (PAI-2) tienen la mayor importancia fisiológica.
Ariel Castro Santana 85833
ResponderEliminarActivación plaquetaria
Un enorme número de sustancias puede actuar sobre las plaquetas e inducir su activación [el factor de activación plaquetaria,6 trombina, el tromboxano (TXA2), fosfato de adenosina (ADP),10 epinefrina, vasopresina y serotonina] a través de sus respectivos receptores, pero todas lo hacen a merced de rutas de señalización intracelular que involucran un sistema de proteínas G acoplado a enzimas efectoras comolas fofolipasas A2 (PLA2) y C (PLC) y adenilato y guanilato ciclasas.5, 6, 10
Hasta ahora se han identificado nueve componentes diferentes de la familia de proteínas G en las plaquetas y, dependiendo del tipo de proteína G que participe en la señalización. Se puede activar o inhibir el efecto.6 Un ejemplo lo constituye la regulación por proteína G de la adenilato ciclasa y por tanto de la producción de monofosfato de adenosina cíclico (AMPc). Un incremento de las cantidades de AMPc bloquea la liberación de calcio en el STD e inhibe la función de la plaqueta. Este es el mecanismo que utilizan las prostaciclinas del endotelio vascular sano para inhibir la activación plaquetaria.
DIAPOSITIVA #DOS (2)
Coagulación de la Sangre
Cuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse.
Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas.
Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
Ariel Castro Santana 85833
ResponderEliminarCoagulación de la sangre
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc.
En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares.
A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (preacelerina —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para activarse y funcionar, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
Ariel Castro Santana 85833
ResponderEliminarVía Intrínseca
Recibe este nombre debido a que antiguamente se pensaba que la sangre era capaz de coagular "intrínsecamente" por esta vía sin necesidad de contar con la ayuda de factores externos. Actualmente se sabe que esto no es exactamente así. De hecho la vía extrínseca es la que realmente inicia el proceso y la vía intrínseca sirve de amplificación y seguridad del proceso hemostático.
El proceso de coagulación en esta vía se desencadena cuando la sangre entra en contacto con una superficie "extraña", es decir, diferente al endotelio vascular.
En el caso de una lesión vascular, la membrana basal del endotelio o las fibras colágenas del tejido conectivo, proporcionan el punto de iniciación.
En general las superficies polianiónicas (cargadas negativamente) pueden cumplir el mismo papel, tanto materiales orgánicos como la celulosa, o no orgánicos como el vidrio, el caolín o algunas resinas pueden actuar como desencadenantes de la reacción.
A esta vía es posible subdividirla en tres pasos:
-Formación del factor XIa
En esta etapa participan cuatro proteínas: Precalicreína, Quininógeno de alto peso molecular (HMWK) y los factores XII y XI. Esta etapa no requiere de iones calcio.
Estos cuatro factores se adsorben sobre la superficie cargada negativamente, formando el complejo cebador o de iniciación. De estos factores el XII funciona como verdadero iniciador, ya que si bien es una proenzima, posee una pequeña actividad catalítica que alcanza para activar a la precalicreína convirtiéndola en calicreína.
En segunda instancia la calicreína actúa catalíticamente sobre el factor XII para convertirlo en XIIa, una enzima muchísimo más activa. La actividad catalítica de la calicreína se ve potenciada por el HMWK.
Por último la proteasa XIIa actúa sobre el factor XI para liberar XIa.
-Formación del factor IXa
El factor IX se encuentra en el plasma como una proenzima. En presencia de iones Ca2+ el factor XIa cataliza la ruptura de una unión peptídica en la molécula del factor IX para formar un glucopéptido de 10 KDa y liberar por otro lado al factor IXa.
El factor IX se encuentra ausente en personas con hemofilia tipo B.
-Formación del factor Xa
Sobre la membrana de las plaquetas se forma un complejo constituido por los factores IXa, X y VIII.
Los residuos gamma-carboxiglutamato de los factores IXa y X actúan como quelantes del ion Ca2+, permitiendo que estos componentes formen un complejo unido por medio de puentes de iones calcio y ayudando a que el complejo se ancle a los fosfolípidos de membrana.
Primero se unen los factores X y IXa a la membrana y luego se une el VIII.
El factor VIII es en realidad un homorodímero, formado por cuatro cadenas proteicas, cada una codificada por un gen diferente (VIII:C y VIII:R). El componente VIII:C es conocido como "componente antihemofílico" y actúa como cofactor del IXa en la activación del factor X, el componente VIII:R es el que permite la unión del factor VIII al complejo.
La ausencia del componente antihemofílico causa hemofilia A.
El complejo formado por los factores IXa-X-VIII-Fosfolípidos y Ca2+ actúa sobre el factor X para convertirlo en Xa.
En este punto concluye la vía intrínseca.
Ariel Castro Santana 85833
ResponderEliminarTrombo & Cuagulo
Los coágulos sanguíneos son masas que se presentan cuando la sangre se endurece pasando de líquida a sólida.
Un coágulo sanguíneo que se forma dentro de una de las venas o las arterias se denomina trombo y también se puede formar en el corazón.
Un trombo que se desprende y viaja desde un lugar en el cuerpo a otro se llama émbolo.Un trombo o émbolo puede bloquear parcial o totalmente el flujo de sangre en un vaso sanguíneo.
Una obstrucción en una arteria puede impedir que el oxígeno llegue a los tejidos en esa área, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede provocar daños en los tejidos o la muerte.
Una obstrucción en una vena generalmente provocará acumulación de líquido e hinchazón.
Las situaciones en las que es más probable que se forme un coágulo de sangre abarcan:
Estar en reposo en cama por largo tiempo.
Cruzar las piernas durante largos periodos al estar sentado o sentarse durante mucho tiempo, como en un avión o en un vehículo.
Durante y después del embarazo.
No tener suficiente agua en el cuerpo (deshidratación).
Tomar píldoras anticonceptivas u hormonas estrógeno (especialmente en las mujeres que fuman).
Utilizar un catéter intravenoso por largo tiempo.
Los coágulos de sangre también son más probables en personas con cáncer, cirugía o lesión recientes, obesidad y enfermedades del hígado o del riñón.
Una acumulación de colesterol que estreche una arteria puede cambiar o reducir el flujo de sangre, facilitando la formación de un coágulo sanguíneo o de un trombo.
Las afecciones que se transmiten de padres a hijos (hereditarias) pueden hacerlo más propenso a la formación de coágulos sanguíneos anormales. Los trastornos hereditarios que afectan la coagulación son:
Trombofilia por factor V de Leiden
Mutación de la protrombina G20210A
Otras afecciones raras como deficiencias de antitrombina III, proteína C y S
Un coágulo de sangre puede bloquear una arteria o una vena en el corazón, afectando:
El corazón (angina de pecho o un ataque cardíaco)
Los intestinos (isquemia mesentérica) o trombosis venosa mesentérica
Los riñones (trombosis de la vena renal)
Las arterias de las piernas o brazos
Las piernas (trombosis venosa profunda)
Los pulmones (embolia pulmonar)
El cuello o el cerebro (accidente cerebrovascular)
Los trombos y émbolos pueden fijarse firmemente a un vaso sanguíneo y pueden bloquear parcial o completamente el flujo de sangre a dicho vaso.
Un bloqueo en el vaso sanguíneo impide que el flujo normal de sangre y oxígeno llegue a los tejidos en ese sitio, lo cual se denomina isquemia. Si la isquemia no se trata oportunamente, puede ocasionar daño tisular o necrosis en esa área.
Ariel Castro Santana 85833
ResponderEliminarFibrinólisis
Después de que el coágulo se ha establecido, comienza la reparación de los tejidos afectados con el proceso de cicatrización. Para hacer posible esto el coágulo es colonizado por células que formarán nuevos tejidos y en el proceso va siendo degradado.
La degradación de la fibrina (fibrinólisis), componente mayoritaria del coágulo, es catalizada por la enzima plasmina, una serina proteasa que ataca las uniones peptídicas en la región triple hélice de los monómeros de fibrina.
La plasmina se genera a partir del plasminógeno, un precursor inactivo; activándose tanto por la acción de factores intrínsecos (propios de la cascada de coagulación) como extrínsecos, el más importante de los cuales es producido por el endotelio vascular. Se le denomina "activador tisular del plasminógeno" (t-PA).
El gen de este factor ha sido clonado y actualmente se puede obtener la proteína producida por tecnología de ADN recombinante.
Este factor suele utilizarse en clínica para favorecer la disolución de trombos.
DIAPOSITIVA #OCHO (8)
Sistema Fibrinolitico
La fibrinolisis consiste en la degradación de las redes de fibrina formadas en el proceso de coagulación sanguínea, evitando la formación de trombos.
La plasmina en su forma activa es la encargada de la degradación de las redes de fibrina, que pasarán a ser fibrinopéptidos solubles tras la fibrinolisis. Estos productos de degradación de la fibrina (PDF), como el Dímero-D, son eliminados normalmente por proteasas en los macrófagos del hígado y el riñón.
La activación de plasmina a partir de plasminógeno ocurre a través de dos vías, la extrínseca y la intrínseca:
En la vía extrínseca, que es estimulada por situaciones como el descenso de la presión parcial de oxígeno en sangre o las infecciones bacterianas, se produce una segregación de diversas sustancias que posibilitarán la activación del plasminógeno para convertirse en plasmina. Dichas sustancias, segregadas por el endotelio, son la uroquinasa y el activador tisular del plasminógeno o tPA.
En la vía intrínseca es la calicreína (KK) la encargada de mediar la activación del plasminógeno.
La fibrinolisis se encuentra regulada por dos factores inhibitores principales: la alfa 2-antiplasmina, que imposibilita la formación de plasmina inhibiendo la activación del plasminógeno, y el inhibidor de tPA, que actúa en la vía extrínseca evitando que el tPA posibilite la activación del plasminógeno.
Dariana Japa 85113
ResponderEliminarLa fibrinólisis es un proceso que previene los coágulos de sangre de crecimiento y convertirse en problemática [1] Este proceso tiene dos tipos:. Fibrinolisis primaria y fibrinólisis secundaria. El tipo principal es un proceso normal del cuerpo, mientras que la fibrinólisis secundaria es la descomposición de los coágulos debido a un medicamento, un trastorno médico, o alguna otra causa.
En la fibrinólisis, un coágulo de fibrina, el producto de la coagulación, se descompone. [2] Su plasmina principal enzima corta la malla de fibrina en varios lugares, que conduce a la producción de fragmentos circulantes que se eliminan por otras proteasas o por el riñón y el hígado .
La plasmina se produce en una forma inactiva, el plasminógeno, en el hígado. Aunque plasminógeno no puede escindir la fibrina, que todavía tiene una afinidad por ella, y se incorpora en el coágulo cuando se formó.
Activador del plasminógeno tisular (t-PA) [3] y uroquinasa son los agentes que convierten el plasminógeno en la plasmina activa, permitiendo de este modo que se produzca la fibrinólisis. t-PA se libera en la sangre muy lentamente por el endotelio dañado de los vasos sanguíneos, de tal manera que, después de varios días (cuando el sangrado se ha detenido), el coágulo se descompone. Esto ocurre porque plasminógeno se convirtió en atrapado dentro del coágulo cuando se formó, ya que se activa lentamente, se descompone la malla de fibrina. t-PA y uroquinasa se sí mismos inhibidas por el inhibidor del activador del plasminógeno-1 y el inhibidor del activador del plasminógeno-2 (PAI-1 y PAI-2). En contraste, la plasmina estimula aún más la generación de plasmina mediante la producción de formas más activas de tanto activador del plasminógeno tisular (tPA) y la uroquinasa.
Alfa 2-antiplasmina y alfa 2-macroglobulina inactivan plasmina. Actividad de la plasmina también se reduce por el inhibidor de la fibrinólisis activable por trombina (TAFI), que modifica la fibrina para que sea más resistente a la del plasminógeno mediada por tPA.
Massiel Rosario 89427.
ResponderEliminarActivacion Plaquetaria: Las plaquetas o trombocitos son fragmentos citoplasmáticos pequeños, irregulares y carentes de núcleo, de 2-3 µm de diámetro,1 derivados de la fragmentación de sus células precursoras, los megacariocitos; la vida media de una plaqueta oscila entre 8 y 12 días. Las plaquetas juegan un papel fundamental en la hemostasia y son una fuente natural de factores de crecimiento. Estas circulan en la sangre de todos los mamíferos y están involucradas en la hemostasia, iniciando la formación de coágulos o trombos. Las plaquetas liberan un gran número de factores de crecimiento incluyendo el factor de crecimiento derivado de plaquetas (PDGF, por platelet derived growth factor), un potente agente quimiotáctico, y el factor de crecimiento transformante beta, (TGF-beta, por transforming growth factor) el cual estimula el depósito de matriz extracelular; Estos dos factores de crecimiento han demostrado jugar un papel significativo en la regeneración y reparación del tejido conectivo; Otros factores de crecimiento producidos por las plaquetas y asociados a los procesos curativos incluyen: factor de crecimiento básico del fibroblasto (basic fibroblast growth factor), factor de crecimiento-1 asociado a la insulina (IGF-1 del inglés insulin-like growth factor-1), factor de crecimiento del epitelio (EGF del inglés epithelial growth factor), factor de crecimiento del hepatocito (HGF del inglés hepatocyte growth factor) y el factor de crecimiento del endotelio vascular (VEGF del inglés vascular endothelial growth factor). La aplicación local de estos factores de crecimiento en altas concentraciones a través del plasma rico en plaquetas (PRP del inglés platelet-rich plasma) ha sido utilizada, por varias décadas, para acelerar el proceso curativo de diferentes lesiones.3 4 5 6 7 8 9.
La función plaquetaria consiste en el mantenimiento del corazón; Esto es alcanzado primariamente por la formación de trombos, cuando existe lesión del endotelio de los vasos sanguíneos. Por el contrario, la formación de trombos es inhibida en el caso de no existir daño en el endotelio.
La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria(plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
Las plaquetas activadas cambian su forma haciéndose más esféricas, y formando pseudopodos en su superficie. De esta forma toman una forma estrellada.
ACtivacion plaquetaria
ResponderEliminarLa superficie interna de los vasos sanguíneos está revestida por una capa delgada de células endoteliales las cuales en circunstancias normales actúan inhibiendo la activación plaquetaria mediante la producción de monóxido de nitrógeno, ADPasa endotelial, y PGI2; la ADPasa endotelial despeja la vía para la acción del activador plaquetario ADP.
Las células endoteliales producen una proteína llamada factor de von Willebrand (FvW),un ligando que media la adhesión celular, el cual ayuda a las células endoteliales a adherir el colágeno a la membrana basal; en condiciones fisiológicas, el colágeno no está expuesto al flujo sanguíneo; el FvW es secretado esencialmente en el plasma por las células endoteliales, y almacenado en gránulos dentro de las células endoteliales y plaquetas.
Cuando la capa endotelial es lesionada, el colágeno, el FvW y el factor tisular del endotelio son expuestos al flujo sanguíneo.
Cuando las plaquetas hacen contacto con el colágeno o el FvW, son activadas; estas son activadas también por la trombina (formada con la ayuda del factor tisular). También pueden ser activadas por una superficie cargada negativamente, como el vidrio.
La activación plaquetaria posterior resulta en el transporte mediado por la escramblasa, de fosfolípidos cargados a la superficie plaquetaria(plaquetas); estos fosfolípidos proporcionan una superficie catalítica (con la carga provista por la fosfatidilserina y fosfatidiletanolamina) para los complejos tenasa y protrombinasa. Los iones de calcio son esenciales para la activación de los factores de coagulación.
Coagulacion de la sangre
EliminarCuando una lesión afecta la integridad de las paredes de los vasos sanguíneos, se ponen en marcha una serie de mecanismos que tienden a limitar la pérdida de sangre. Estos mecanismos llamados de "hemostasia" comprenden la vasoconstricción local del vaso, el depósito y agregación de plaquetas y la coagulación de la sangre.
Se denomina coagulación al proceso por el cual la sangre pierde su liquidez, tornándose similar a un gel en primera instancia y luego sólida, sin experimentar un verdadero cambio de estado.
Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble y con la capacidad de entrelazarse con otras moléculas iguales, para formar enormes agregados macromoleculares en forma de una red tridimensional.
El fibrinógeno, una vez transformado, recibe el nombre de fibrina. La coagulación es por lo tanto, el proceso enzimático por el cual el fibrinógeno soluble se convierte en fibrina insoluble, capaz de polimerizar y entrecruzarse.
Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas.
Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.
Factores de coagulación
El proceso de coagulación implica toda una serie de reacciones enzimáticas encadenadas de tal forma que actúan como un alud o avalancha, amplificándose en cada paso: un par de moléculas iniciadoras activan un número algo mayor de otras moléculas, las que a su vez activan un número aún mayor de otras moléculas, etc.
En esta serie de reacciones intervienen más de 12 proteínas, iones de Ca2+ y algunos fosfolípidos de membranas celulares.
A cada uno de estos compuestos participantes en la cascada de coagulación se les denomina "Factor" y comúnmente se lo designa por un número romano elegido de acuerdo al orden en que fueron descubiertos.
Siete de los factores de coagulación (precalicreína —factor V—, protrombina —Factor II—, proconvertina —factor VII—, factor antihemofílico beta —IX—, factor Stuart —X—, tromboplastina plasmática —XI— y factor Hageman —XII—) son zimógenos sintetizados en el hígado, esto es, proenzimas que normalmente no tienen una actividad catalítica importante, pero que pueden convertirse en enzimas activas cuando se hidrolizan determinadas uniones peptídicas de sus moléculas.
Estas proenzimas, una vez recortadas, se convierten en proteasas de la familia de las serina proteasas; capaces de activar a las siguientes enzimas de la cascada.
Una enzima activa "recorta" una porción de la siguiente proteína inactiva de la cascada, activándola.
Algunos factores de coagulación requieren vitamina K para su síntesis en el hígado, entre ellos los factores II (protrombina), VII (proconvertina), IX (antihemofílico beta) y X (Stuart).
Este comentario ha sido eliminado por un administrador del blog.
EliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminararislendy castillo 2013-0654
ResponderEliminar1. La coagulación comienza en respuesta a una lesión en un vaso sanguíneo. Es un proceso donde se producen una serie de reacciones en cadena de varios tipos celulares y proteínas para formar un coágulo evitando asi la pérdida excesiva de sangre. Un coágulo consiste en una red de proteínas insolubles como la fibrina con plaquetas y células atrapadas que bloquea la salida de sangre hasta que se repare el tejido.
2. En la fase de iniciación es fundamental la formación de pequeñas cantidades de trombina. La lesión expone las proteínas solubles de la sangre a tejidos circundantes y a la matriz extracelular. Estos genera una serie de reacciones que tiene lugar cuando el factor VIIa se une a los factores tisulares en las membranas de las células que están fuera de los vasos originando el complejo VIIa/TF. Activa a su vez pequeñas cantidades de factor IX y X. El factor Xa se asocia con el factor Va en la superficie de las células que presentan los factores tisulares.
3. Se forma un complejo protrombinasa que da origen a pequeñas cantidades de trombina (factor IIa). El factor V puede ser activado por factor Xa, por proteasas de la matriz extracelular o ser liberado de las plaquetas al contactar con el colágeno u otros componentes de la matriz extracelular.
En la fase de amplificación se produce la activación de las plaquetas por la trombina generada en la fase de iniciación. Factores en la superficie de las plaquetas provocan la producción masiva de trombina. Tiene lugar sólo si la lesión permite la salida de plaquetas y del complejo VIII/vWF. Se secretan formas parcialmente activadas de factor V en la superficie. Se disocia el complejo VIII/vWF, permitiendo al factor vWF mediar la adhesión y agregación adicional de plaquetas y al factor VIII activarse y unirse a la membrana de las plaquetas. La trombina activa el factor XI en la superficie de la plaqueta durante esta fase.
En la fase de propagación se produce trombina de forma masiva para la formación de un coágulo estable. El factor IXa, generado en la fase de iniciación, se une al factor VIIIa en la superficie de las plaquetas. El factor XIa en la superficie de plaquetas permite la unión de más cantidad de factor IXa. El factor Xa generado por el complejo IXa/VIIIa formado en la membrana de las plaquetas se une con el factor Va. Al ensamblarse la protrombinasa (Xa/Va) provoca una masiva producción de trombina en cantidad suficiente para que se forme el coágulo.